
Abstract
The number of nephrons is a multifactorial trait controlled by the interaction of environmental factors and genetic variants that influence the extent of branching nephrogenesis during foetal life. A correlation between renal mass and nephron number in newborns allows the use of the total kidney volume at birth as a surrogate for congenital nephron number. Since the renin–angiotensin system plays an important role in renal development we hypothesized that the common, functional insertion/deletion (I/D) polymorphism in the ACE gene might be responsible for the variation in kidney size amongst healthy individuals. We recruited 210 healthy Polish full-term newborns born to healthy women with uncomplicated pregnancies. The kidney volume was measured sonographically. Total kidney volume (TKV) was calculated as the sum of left kidney volume and right kidney volume. TKV was normalized to body surface area (TKV/BSA). The I and D alleles were identified using polymerase chain reaction. TKV/BSA in newborns carrying at least one insertion ACE allele was significantly reduced by approximately 8% as compared with homozygous newborns for the D allele (DD genotype) (105.1±23.6 vs. 114.2±28.2 cm3/m2, p<0.05). The results of this study suggest that I/D ACE polymorphism may account for subtle variation in kidney size at birth, which reflects congenital nephron endowment.
Introduction
The kidney is a key organ in the development of hypertension. In 1988, Brenner et al. postulated that a reduced number of nephrons contributes to essential hypertension. 1 In 2003, Keller et al. provided evidence for Brenner’s hypothesis. 2 Using a three-dimensional stereological method the authors reported that patients with hypertension had significantly fewer glomeruli per kidney than matched normotensive controls. Patients with hypertension also had a significantly greater glomerular volume than did the controls but very few obsolescent glomeruli. 2
Despite extensive research, still little is known about how final nephron number is set. 3 However, genetic studies in humans and mice have provided valuable insights into genetic contribution and molecular mechanisms leading to normal nephron endowment and renal hypoplasia. 4 In addition, El Kares et al. recently hypothesized that congenital nephron number is a multifactorial trait controlled by the interaction of environmental factors and genetic variants that influence the extent of branching nephrogenesis during foetal life. 5 In humans, final nephron endowment for life is determined during late gestation and displays wide individual variation.6-8 In addition, confirmation of a strong correlation between renal mass and nephron number in newborns allowed establishment of total kidney volume at birth (measured by ultrasonography) as a surrogate for congenital nephron number. 9
Tufro-McReddie et al. demonstrated that blockade of angiotensin II type 1 receptors (AT1) during the late nephrogenic period in rats induced an arrest in nephrovascular maturation and renal growth, resulting in altered kidney architecture characterized by fewer, thicker, shorter afferent arterioles, reduced glomerular size and number, and tubular dilation. In addition, the inhibition of angiotensin I-converting enzyme (ACE) in tadpoles (Rana catesbeiana) induced even more marked developmental renal abnormalities with reduction of nephron number. 10 These findings clearly indicate that an intact renin–angiotensin system (RAS) is a prerequisite for normal renal development. 11
The ACE gene contains a polymorphism based on the presence (insertion (I)) or absence (deletion (D)) within intron 16 of a 287-base-pair (bp) DNA fragment, resulting in three genotypes (II and DD homozygotes, and ID heterozygotes). 12 This polymorphism is reported to be associated with approximately 28% of the variation in plasma ACE levels, depending on the ethnic background examined. 13 Both plasma ACE level and renal ACE expression are highest in subjects with DD genotype, intermediate in ID heterozygotes and lowest in II homozygotes.13,14
We therefore hypothesized that this common polymorphism in the ACE gene might also be responsible for the variation in nephron number that is seen amongst healthy individuals. To verify this hypothesis, we examined association of the I/D ACE polymorphism with congenital kidney volume, a surrogate measure of the number of nephrons, in a cohort of healthy, full-term newborns in Poland.
Materials and methods
Study subjects
At the Department of Neonatal Diseases at the Pomeranian Medical University in Szczecin we prospectively recruited 210 consecutive healthy Polish newborns (96 females and 114 males), born after the end of the 37th week of gestation to healthy women with uncomplicated pregnancies. All children were breast-fed and free of medication. Twins, infants of mothers with preeclampsia, hypertension of any cause, diabetes, history of illicit substance use or antenatal steroid therapy were excluded. Other exclusion criteria were congenital infection, intra-uterine growth restriction (i.e. below the 10th percentile birth mass, length or head circumference), chromosomal aberrations or congenital malformations. At birth, cord blood of newborns was obtained for isolation of genomic DNA. The gender of the newborn, birth mass (BM) and birth length (BL) were taken from standard hospital records. Body surface area (BSA) was calculated as the square root of [BL (cm) × BM (kg)/3600] according to Mosteller. 15 The study was approved by the local ethics committee and parents gave informed consent.
Kidney volume measurement
Sonographic examination was performed by an ultrasound specialist (AK) in newborns on the third day after delivery with an EnVisor C machine (Philips Canada, Markham, Ontario, Canada) using a 5 MHz sector probe (Philips, Canada) and 10 MHz linear probe (Philips, Canada) as described previously.16,17 Left and right kidney volumes (LKVs and RKVs, respectively) were calculated using the following formula for volume of an ellipsoid: [kidney volume = 4/3Π (length/2)(height/2)(width/2)]. 3 Total kidney volume (TKV) was calculated as the sum of LKV and RKV. Subsequently, TKV was normalized for body surface area (TKV/BSA).3,16
Determination of ACE genotypes
Genomic DNA from cord blood was isolated using the QIAamp Blood DNA Mini Kit (QIAGEN, Hilden, Germany). The I and D alleles were identified on the basis of PCR amplification of the respective fragments from intron 16 of the ACE gene, and subsequent amplicon size fractionation and visualization by agarose-gel electrophoresis, according to the method introduced by Lindpaintner et al. 18
Statistical analysis
Possible divergence of ACE genotype frequencies from Hardy–Weinberg equilibrium was assessed using a χ2 test. The distribution of each quantitative variable was tested for skewness. Quantitative data were presented as means ± standard deviation (SD). Association of either gender or ACE genotype (with respect to dominant and recessive modes of inheritance of the D allele) with each outcome variable was assessed by Student’s-t test using STATISTICA (StatSoft, Inc. (2011), version 10). Statistical significance was defined as p<0.05.
Results
All PCR samples were genotyped twice and a concordance rate of 100% was attained. There were 57 II ACE homozygotes (27.2%), 100 ID heterozygotes (47.6%) and 53 DD homozygotes (25.2%). The frequency of the deletion allele was 49.0% (49.1% in newborn males and 49.0% in newborn females). The ACE genotype distribution conformed to the expected Hardy–Weinberg equilibrium (p=0.581). Characteristics of the newborn cohort (n=210) with respect to gender are shown in Table 1. The distribution of these characteristics in our cohort, including TKV/BSA, approached normality (skewness <2 for all variables) (Figure 1). The mean values of birth mass, BSA, LKV, RKV and TKV in male newborns were significantly higher as compared with female newborns. No significant differences in gestational age, birth length, LKV/RKV or TKV/BSA were found between male and female newborns.
Characteristics of newborn cohort with respect to gender.

Males versus females. BM: birth mass; BL: birth length; BSA: body surface area; LKV: left kidney volume; RKV: right kidney volume; TKV: total kidney volume.
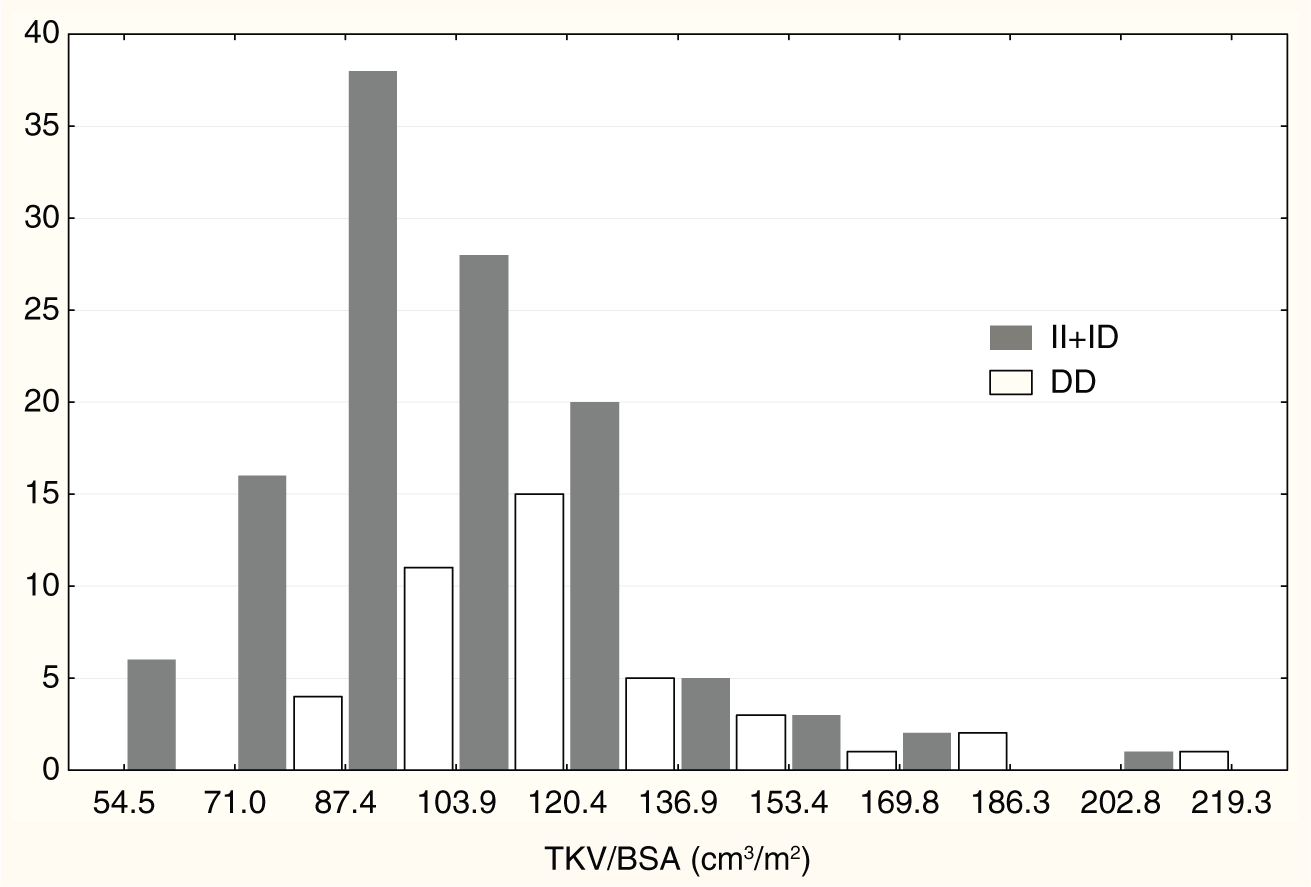
Distribution of TKV/BSA with respect to ACE polymorphism.
Characteristics of kidney volumes with respect to I/D ACE polymorphism are shown in Table 2. The mean values of LKV, RKV, TKV and TKV/BSA in DD homozygotes were significantly higher as compared with newborns with at least one I ACE allele (II+ID, recessive mode of the D allele inheritance) (13.7±3.8 vs. 12.5±3.1 cm3, 13.0±3.4 vs. 11.6±3.2 cm3, 26.7±6.9 vs. 24.1±5.7 cm3, 114.2±28.2 vs. 105.1±23.6 cm3/m2, respectively). No significant differences in LKV/RKV ratio were found between newborns with different ACE genotypes. No significant differences in kidney volumes were found between newborns with at least one D ACE allele (ID+DD) and II homozygotes (dominant mode of the D allele inheritance).
Characteristics of the kidney volumes in newborns with respect to ACE polymorphism.
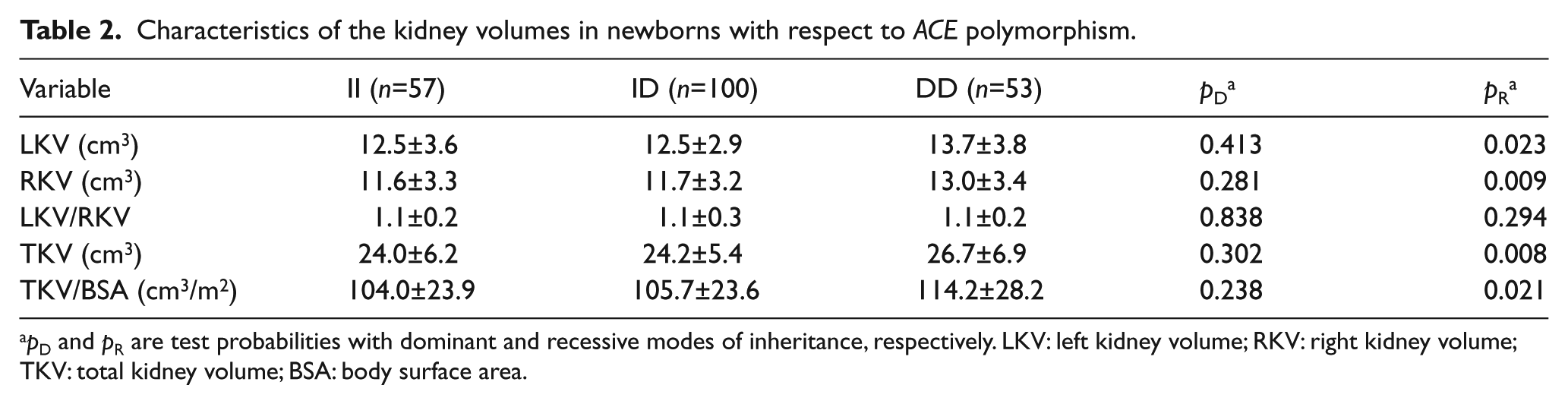
pD and pR are test probabilities with dominant and recessive modes of inheritance, respectively. LKV: left kidney volume; RKV: right kidney volume; TKV: total kidney volume; BSA: body surface area.
Discussion
The present study demonstrates for the first time an association between the I/D ACE polymorphism and kidney size in newborns born full term. TKV/BSA was reduced in newborns carrying at least one insertion ACE allele, as compared with DD homozygous newborns. This suggests that the I/D ACE polymorphism may, analogously with other common polymorphisms in developmental genes including PAX2, 3 RET, 9 ALDH1A2 5 and OSR1, 19 partially account for subtle variation in kidney size at birth, which reflects congenital nephron endowment. In accordance with Brenner’s hypothesis, newborns having an unfavourable polygenic profile associated with reduced nephron number may benefit from early and aggressive surveillance for essential hypertension in adulthood. 1
RAS plays a critical role in kidney development.11,20 Loss-of-function mutations in the genes encoding components of the RAS or pharmacological inhibition of RAS in animals or humans cause diverse renal malformations,21-28 including low nephron endowment.10,29,30 However, the mechanisms by which an intact RAS controls proper renal system development, and how decreased angiotensin II (ANG II) generation or availability results in abnormal kidney development are still not fully elucidated. 31
Human kidney development begins at approximately the fifth week of gestation, when metanephric mesenchymal cells secrete glial-derived neurotrophic factor (GDNF), which interacts with the c-RET tyrosine kinase receptor expressed in the ureteric bud (UB) tip cells to induce branching. 32 In response to the trophic effects of GDNF, a ureteric bud sprouts from each nephric duct and arborizes within the lateral mesenchyme. 9 Signals from each ureteric bud branch tip induce adjacent metanephric stem cells to form individual nephrons, which fuse to the tree-like collecting system. It is noteworthy that the deficit of nephron number results from a defect in the branching morphogenesis of the ureteric bud and even subtle decreases in the efficiency of UB branching result in a profound decrease in nephron endowment (number). 33 ANG II is emerging as a stromal factor that regulates UB morphogenesis. Stimulation of its type 1 receptor by ANG II inhibits Spry1 gene expression, and thereby relieves inhibition of signalling via the Gdnf/c-Ret/Wnt11 pathway. In addition, coupling of ANG II with its type 2 receptor upregulates paired box 2 (Pax2) and stimulates UB branching. 31
In humans, renal ACE is detected at approximately 30 days of gestation, and is localized to proximal tubules in increasing amounts until birth. 34 Mizuiri et al. reported that the number of ACE transcripts in 100 ng of total renal RNA was significantly lower in adult subjects with the II genotype compared with ID heterozygotes or DD homozygotes. 14 On the other hand, the intron 16 ACE polymorphism does not seem to directly affect regulation of ACE gene expression, and the mechanism causing ACE level variability should be referred to another locus in strict linkage disequilibrium with the I/D polymorphism. However, to date, such a functional variant in the ACE gene has not been unequivocally identified.35-37 The relationship between I/D ACE polymorphism and ANG II level seems to be less clear. However, Abraham et al. found that not only plasma ACE activity but also plasma ANG II concentration are the highest in DD homozygotes. 38 In addition, Ueda et al. documented significantly increased ANG II generation in response to ANG I infusion in normotensive male DD homozygotes. 39 Therefore, we regard as plausible the claim that the I/D polymorphism of the ACE gene might be considered as a predictor for the rate of ANG II formation14,39 and is therefore important in renal development.
We are aware that potential confounding variables that could create a spurious association might constitute a relevant limitation of our study. Therefore, to minimize their effects, the study was conducted in a carefully selected, fairly homogeneous group of newborns, all of whom met all criteria for inclusion. The sonographic examination of kidney size was performed according to protocols in the literature that give reproducible results, and kidney volumes were similar to those of a large cohort of Danish newborns who were studied within the first five days of life. 16 The TKV/BSA ranges in infants with at least one insertion ACE allele and in DD homozygotes were similar (Figure 1). The ACE genotype distribution in our group was in accordance with Hardy–Weinberg equilibrium, and allele frequencies were similar to those reported in aboriginal-European newborns.40,41 It is therefore very unlikely that our cohort of newborn infants is a biased sample. LKV/RKV ratio did not differ among newborns with the various genotypes. Thus, the association of the I ACE variant with a reduction in TKV cannot be attributed to a few newborns with very small kidneys or with unilateral renal hypoplasia.
In addition, the association of DD ACE genotype with normal kidney size led us to suspect that the theory of ‘antagonistic pleiotropy’ is relevant here. In this theory Williams 42 suggested that some genes responsible for increased fitness in the younger, fertile organism contribute to decreased fitness later in life. Our results suggest that DD homozygosity, usually regarded as deleterious in adults,43,44 might well be advantageous in early human development.
Conclusions
The I/D ACE polymorphism may, as analogously with other common polymorphisms in the developmental genes, account for subtle variation in kidney size at birth reflecting congenital nephron endowment.
Footnotes
Acknowledgements
The authors would like to thank Dr Jeremy Clark at the Department of Clinical and Molecular Biochemistry, Pomeranian Medical University, Szczecin, Poland for his support and valuable comments.
Conflict of interest
There are no competing interests.
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.