Gas phase fragmentation reactions of monoprotonated 4-(3-aminopropyl)- and 4-(4-aminobutyl)-3-hydroxyfurazan were investigated to examine potential interactions between functional groups. The two heterocyclic alkyl amines were ionized by electrospray ionization (ESI, positive mode) and fragmented using tandem mass spectrometry (MS/MS). The fragmentation pathways were characterized using pseudo MS3 experiments, precursor-ion scans, and density functional computations. For both heterocyclic ions, loss of ammonia was the only fragmentation process observed at low collision energies. Computational analysis indicated that the most feasible mechanism was intramolecular nucleophilic displacement of ammonia from the protonated ω-aminoalkyl side chain by N5 of the furazan ring. The alkylated nitrogen in the resulting bicyclic product ion facilitated N-O bond cleavage; subsequent neutral losses of nitric oxide (NO) and carbon monoxide (CO) occurred by homolytic bond cleavages. Next in the multistep sequence, neutral loss of ethylene from a radical cation was observed. A less favorable, competing fragmentation pathway of protonated 4-(3-aminopropyl)-3-hydroxyfurazan was consistent with cleavage of the 3-hydroxyfurazan ring and losses of NO and CO. Overall, the similar fragmentation behavior found for protonated 4-(3-aminopropyl)- and 4-(4-aminobutyl)-3-hydroxyfurazan differed from that previously characterized for furazan analogs with shorter alkyl chains. These observations demonstrate that a small change in the structure of multifunctional, heterocyclic alkyl amines may significantly influence interactions between distinct functional groups and the nature of the fragmentation process.
The rings in the structures of biologically active heterocycles, such as pharmaceuticals,1–3 antifungal agents,4,5 and herbicides,6 provide both a rigid scaffold and hydrogen bonding sites.7–10 When analyzed by tandem mass spectrometry (MS/MS), heterocyclic ions often fragment by a prominent and characteristic ring cleavage11–18 or by cleavage of a bond to the ring.19 In other instances, the ring participates in proton transfer prior to the dissociative step and is located intact in the neutral product or the product ion.20–24 In general, distinctive gas phase, MS/MS fragmentation processes are valuable for the identification and structure elucidation of trace amounts of substances as well as for quantitative determinations using selected or multiple reaction monitoring (SRM/MRM) techniques.25,26
Heterocycles containing a hydroxy substituent and three contiguous heteroatoms in a five-membered ring have been recognized as bioisosteres of the carboxyl group and then utilized as a drug design strategy.7,8,27–36 Accordingly, 3-hydroxyfurazan heterocycles have been incorporated into the structures of glutamate analogs,28,32 dihydroorotate dehydrogenase inhibitors,30 as well as anticancer33,35 and immunosuppressive agents.34
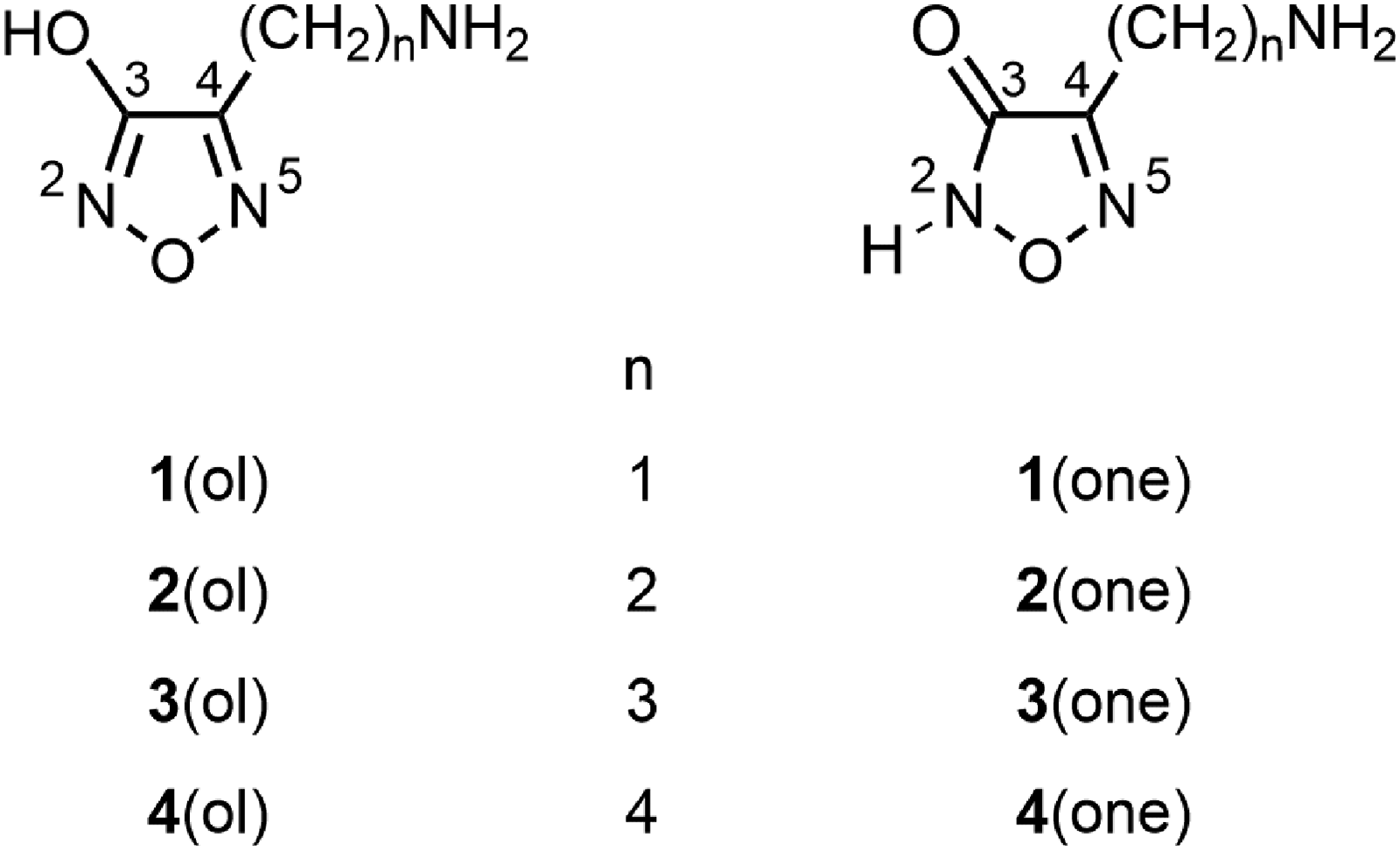
The aminomethyl- and 2-aminoethyl 3-hydroxyfurazans 1 and 2 were readily protonated by electrospray ionization mass spectrometry (ESI MS, positive mode) and fragmented by ring cleavage upon collision-induced dissociation (CID).37 In the lowest energy pathway, ring cleavage was accompanied by neutral losses of nitric oxide (NO) and carbon monoxide (CO). At higher collision energies, other fragmentation pathways, including an initial loss of ammonia, were observed as well. The three nitrogen atoms in 4-(ω-aminoalkyl)-3-hydroxyfurazans 1a(ol) and 2a(ol) are potential protonation sites, and the heterocycles may also exist as the higher energy tautomeric, lactam forms 1a(one) and 2a(one). Computational analyses were used to associate the initial step of each pathway with a particular tautomer protonated at a specific site. For example, protonation at N5 led to ring cleavage and successive losses of NO and CO.
In the current investigation, the fragmentation pathways of protonated 4-(3-aminopropyl)- and 4-(4-aminobutyl)-3-hydroxyfurazans (3a and 4a, respectively, Figure 1) were studied using tandem mass spectrometry and density functional theory (DFT) computations. Both cations contain two different functional groups linked by an alkyl chain. Either the primary amine or a tautomer of the heterocycle may be protonated in the gas phase37 and fragmentation may or may not occur through intramolecular interactions. Unlike the fragmentation by furazan ring cleavage documented for protonated 4-(aminomethyl)- and 4-(2-aminoethyl)-3-hydroxyfurazan (1a and 2a, respectively, Figure 1),37 the prominent fragmentation process found for the homologous ions 3a and 4a was the loss of ammonia. This favorable process was attributed to intramolecular displacement of the protonated primary amino group by a nitrogen atom in the ring. Ring cleavage occurred as a secondary process for both protonated furazans and as a competing fragmentation process of 3a at higher collision energies.
Tautomeric 4-(ω-aminoalkyl)-substituted 3-hydroxyfurazans (1,2,5-oxadiazol-3-ols) and 1,2,5-oxadiazol-3(2H)-ones. The corresponding [M + H]+ ions are designated as 1a–4a (mixture of tautomers) or specifically as lactim tautomers, 1a(ol)–4a(ol), and lactam tautomers, 1a(one)–4a(one).
Materials and methods
The 4-(ω-aminoalkyl)-3-hydroxyfurazans (3 and 4) were available from previous studies.27 The hydrochloride salts of 3 and 4 were dissolved in aqueous methanol (1 mg mL–1) and a portion (5–10 μL) was introduced into the spectrometer by flow injection (H2O:MeOH: 1:1, v/v, 20 μL min–1).
Gas phase ions were generated by positive-ion electrospray (ESI(+)) and mass spectra were acquired on Thermo-Finnigan LCQ-DUO ion trap and Micromass Quattro triple quadrupole mass spectrometers. Instrument settings38,39 are provided in the supplemental material (Tables S1 and S2). Pseudo MS3 spectra and precursor-ion scans were acquired after an ion (e.g. 3a) was subjected to non-selective, in-source fragmentation. In-source ions were then mass selected for CID in the ion trap or the collision cell of the triple quadrupole mass spectrometer (MS/MS) to generate pseudo MS3 spectra. In the triple quadrupole mass spectrometer, precursor-ion scans were collected by setting the third quadrupole to select a certain product ion and by scanning ions from the source in the first quadrupole to locate the precursors that dissociate to the selected product ion. Accurate masses were measured on a Bruker Daltonic Compact QToF spectrometer.38
The energetics of fragmentation processes were computed using DFT and Møller-Plesset perturbation theory (MP2) within the Gaussian 09 suite of programs (Revision C.01).40 Geometry optimizations and frequency calculations were performed using the ωB97X-D/6-311 + G(d) functional.41 Energy minima were characterized by having no imaginary vibrational frequencies, whereas saddle points had one such frequency. Thermochemical data are reported as combinations of single-point MP2/6-311++G(2d,p) electronic energies and uncorrected entropies and thermal corrections from the ωB97X-D/6-311 + G(d) calculations and are designated as MP2/6-311++G(2d,p)//ωB97X-D/6-311 + G(d) free energies given in kJ mol–1. Higher energy conformations of a particular ion located computationally are denoted by the addition of a prime to the ion descriptor (e.g. 3aʹ(one) in Figure 4(b)). Cartesian coordinates for the optimized structures are given in the supplemental material.
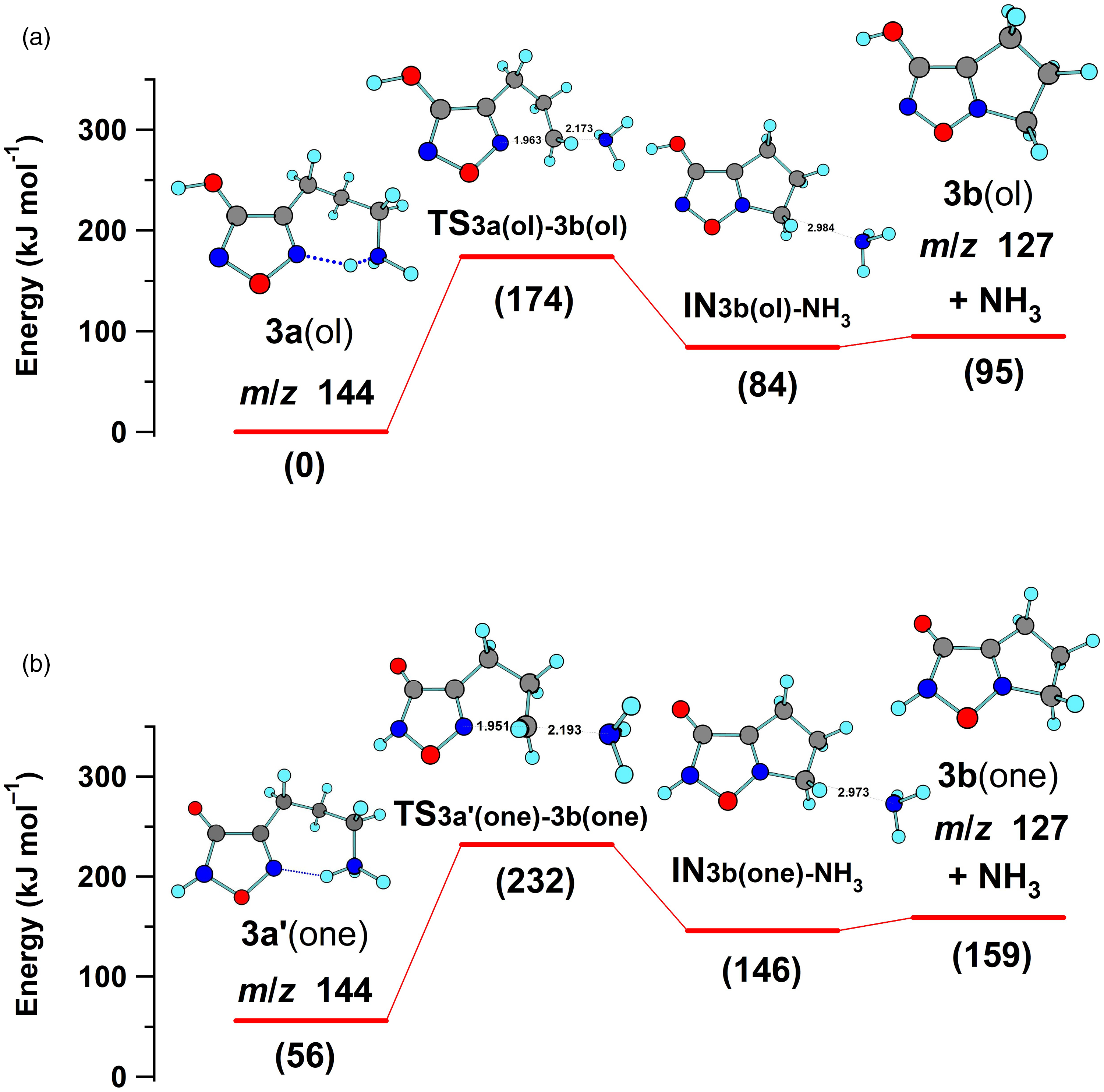
Potential energy profiles computed for the nucleophilic displacement of ammonia from the protonated tautomers of 4-(3-aminopropyl)-3-hydroxyfurazan 3a(ol) (a) and 3a(one) (b).
Results and discussion
The ω-aminoalkyl heterocycles 4-(3-aminopropyl)-3-hydroxyfurazan (3) and 4-(4-aminobutyl)-3-hydroxyfurazan (4) were protonated readily when subjected to ESI. In the ion trap mass spectrometer and at low collision energy (5–10 eV, lab. frame) in the triple quadrupole mass spectrometer, the tandem mass spectra of 3a (m/z 144, Figure 2) and 4a (m/z 158, Figure S1) showed prominent product ions at m/z 127 and m/z 141, respectively, corresponding to a neutral loss of NH3. Accurate mass determinations by MS/MS (m/z 144.0776 → m/z 127.0507 and m/z 158.0921 → m/z 141.0656) confirmed the loss of NH3. At higher collision energies (15–20 eV) in the triple quadrupole mass spectrometer (Figure 2, spectra (d) and (e); Figure S1, spectra D and E), the formation of lower mass product ions (< m/z 110) indicated more extensive fragmentations of 3a and 4a.
Tandem mass spectra of protonated 4-(3-aminopropyl)-3-hydroxyfurazan (3a, m/z 144, [M + H]+) collected on an ion trap mass spectrometer (a) and on a triple quadrupole mass spectrometer (15 V cone) over a range of collision energies (b–e).
Protonation site and initial neutral loss of ammonia
The relative energies of ions 3a and 4a differing in protonation site and conformation were determined computationally (Table S3). Ions protonated at N5 have higher energy and the lowest energy ions 3a(ol) and 4a(ol) were protonated on the primary amino group, the most basic site. The ions were stabilized by intramolecular hydrogen bonds and, typically, conformations of the lactim tautomer were more stable than the corresponding lactam tautomer. Overall, the relative energies of the ions were equivalent to those computed for the shorter chain protonated ω-aminoalkyl-3-hydroxyfurazans (1a and 2a).37 Note that protonation of the primary amino group created structural connections matching the structure of ammonia.37
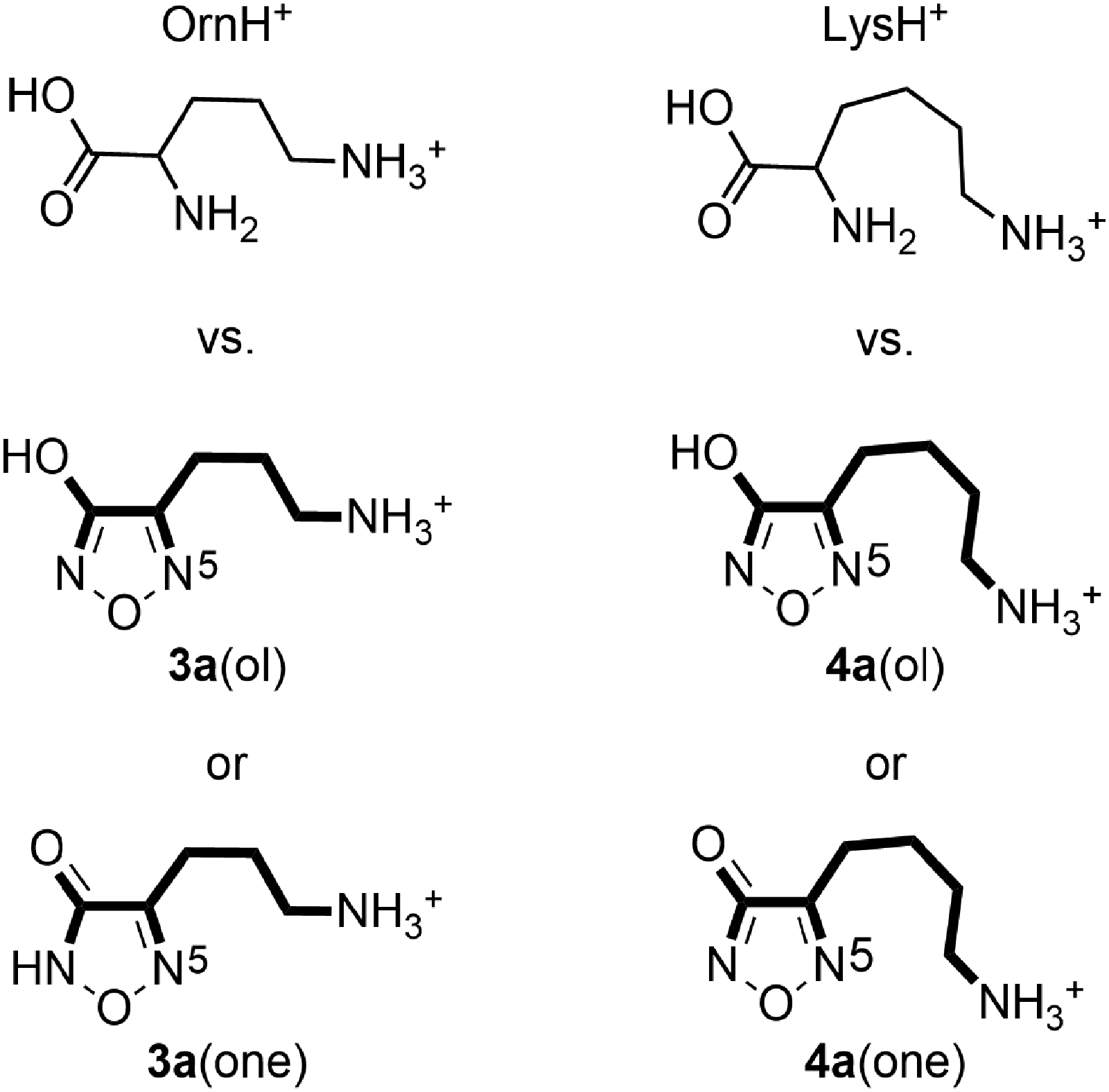
The loss of ammonia from the terminal position of an aliphatic chain has been documented for several protonated, structural analogs, such as diaminoalkanes,42 diamino, dicarboxylic acids,38,42 aminoalcohols,43 muscimol, an aminomethyl substituted hydroxyisoxazole,44–46 and amlodipine, an aminoethoxymethyl substituted dihydropyridine calcium channel blocker.47,48 In particular, the structures of both tautomers of the 3-hydroxyfurazans 3a and 4a closely resemble the structures of the monoprotonated forms of the common, naturally occurring diamino acids ornithine (OrnH+) and lysine (LysH+) (Figure 3). Indeed, the 3-hydroxyfurazan ring is a conformationally constrained analog of the α-amino and carboxyl groups, whereas the ω-aminoalkyl substituents in 3a and 4a are identical to the side chains of ornithine and lysine, respectively. Upon MS/MS, the prominent loss of the side chain nitrogen from OrnH+ and LysH+ was confirmed by isotopic labeling, and computations showed that the nucleophilic displacement of the protonated, side chain amino group by the α-amino group of OrnH+ to form a favorable five-membered ring was feasible.38
Structural comparison of protonated ornithine (OrnH+) with protonated 4-(3-aminopropyl)-3-hydroxyfurazan, 3a(ol), and its tautomer 4-(3-aminopropyl)-1,2,5-oxodiazol-3(2H)-one, 3a(one). The portion of the aminoalkyl heterocycle corresponding to the structure of the diamino acid is highlighted. The analogous structural relationship is found for the protonated homologs lysine (LysH+), 4-(4-aminobutyl)-3-hydroxyfurazan, 4a(ol), and 4-(4-aminobutyl)-1,2,5-oxodiazol-3(2H)-one, 4a(one).
In the current investigation, a reasonable energy (174 kJ mol–1) was computed for the cyclic five-membered transition structure TS3a(ol)-3b(ol) formed upon loss of ammonia from the most stable conformation of 3a(ol) (Figure 4(a)). This displacement of ammonia from the aminopropyl side chain by N5 in the ring of 3a is analogous to the process characterized for OrnH+ (computed barrier = 153 kJ mol–1).38 The potential energy profile was also computed for 3aʹ(one), the higher energy and probably less abundant tautomer in the gas phase. Although the profile was shifted to higher energy (Figure 4(b)), a similar barrier (176 kJ mol–1) was computed for the corresponding N5 nucleophilic displacement of ammonia. Overall, the tautomers 3a(ol) and 3aʹ(one) gave tautomeric, bicyclic product ions (3b(ol) and 3b(one)).
A similar intramolecular, nucleophilic displacement of NH3 from the 4-aminobutyl furazan 4a would form a favorable six-membered ring. The corresponding six-membered ring formation upon the loss of ammonia has been characterized by mass spectrometry for the fragmentation of LysH+,38 which also has a 4-aminobutyl group (Figure 3). With the loss of ammonia as the predominant, initial fragmentation process observed for 4a and LysH+ and the structural correlation of the ions, the evidence is consistent with fragmentation of both ions by the same displacement mechanism.
Although the computations indicate reasonable energetics for the nucleophilic displacement of ammonia, other possible mechanisms were investigated, such as elimination processes. A barrier of 216 kJ mol–1 was computed for the elimination of ammonia from the 3-hydroxyfurazan 3aʹʹ(ol) via abstraction a proton from the propyl side chain by N5 (Figure S2A). For the analogous proton abstraction and elimination of ammonia from the tautomer 3aʹʹʹʹ(one), the potential energy profile (Figure S2B) was shifted to higher energy, but the height of the barrier was similar (230 kJ mol–1). On the other hand, proton abstraction by the exocyclic, carbonyl oxygen required only 157 kJ mol−1 to effect the loss of ammonia from a higher energy conformation (3aʹʹʹ(one)) of the lactam tautomer (Figure S3). The structures of the m/z 127 product ions resulting from these elimination processes differ in the placement of protons on two of three heteroatoms (i.e. N2, N5 and the exocyclic oxygen).
Each of the four mechanisms considered above for the loss of ammonia from 3a required similar inputs of energy but generated a product ion with a distinct structure, namely 3b(ol), 3b(one), 3c(ol), 3c(one), and 3cp. Thus, the secondary fragmentation reactions of 3a were examined to distinguish the ions formed by the initial loss of ammonia and its mechanism.
Fragmentation pathways of 3a
When 3a was subjected to CID at higher collision energies (10–20 eV, lab. frame, MS2) in the triple quadrupole mass spectrometer (Figures 2(c)–2(e)), the primary product ion at m/z 127 was accompanied by major product ions at m/z 69, 58, and 41 and minor product ions at m/z 86 and 85. Precursor-product ion relationships were established for these more extensive fragmentations by pseudo MS3 spectra (Figure S4A), and by precursor-ion scans (Figure S4B). Thus, the fragmentation sequence m/z 144 → 127 → 97 → 69 → 41 was established as the most important pathway of 3a, while the sequence m/z 144 → 86 → 58 → 41 was identified as a competing pathway.
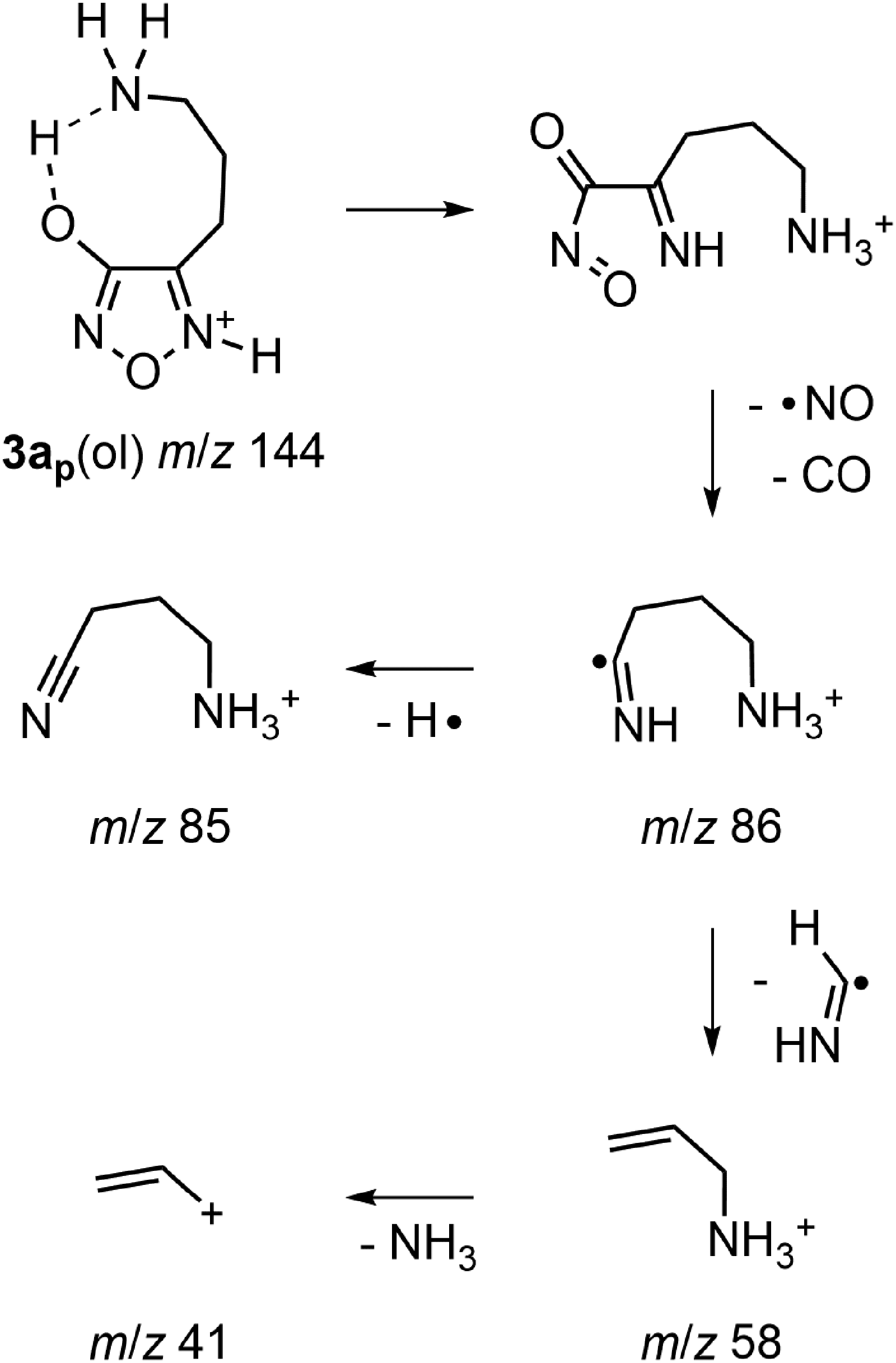
For the competing pathway, the initial neutral loss of 58 u (m/z 144 → 86; Figures 2 and S4B) corresponded to the combined neutral loss of NO and CO. The analogous transition has been characterized as the lowest energy fragmentation process for protonated 4-(aminomethyl)-3-hydroxyfurazan (1a).37 The bond connectivity in the 3-hydroxyfurazan requires ring cleavage to explain the formation of NO and CO. Previous computations on 1a indicated that ring opening by N5–O bond cleavage was achieved with input of modest energy when N5 was protonated and the proton on oxygen was abstracted by the side chain amino group.37 Accordingly, protonation at N5 and abstraction of the oxygen proton by the primary amino group gave the analogous ring opening of 3ap(ol) (Scheme 1). Subsequent homolytic bond cleavage generated NO (a neutral radical), CO and the radical cation (HN = C•–CH2CH2CH2NH3+) observed at m/z 86.
Competing fragmentation pathway of protonated 4-(3-aminopropyl)-3-hydroxyfurazan (3ap(ol), m/z 144) by initial ring cleavage.
In the precursor-ion scans (Figure S4B), the ion at m/z 58 was connected to ions at m/z 86 and 144 (3a), and the ion at m/z 41 was linked to the ion at m/z 58 and to other higher mass ions. Thus, the m/z 86 ion is a likely source of both the ion at m/z 58 (H2C = CHCH2NH3+) and the minor ion at m/z 85 (N≡CCH2CH2CH2NH3+) (Figures 2(c)–2(e) and S4B, middle spectrum) by respective losses of a methanimine radical (HN = CH•) and a hydrogen atom (Scheme 1). The possible conversion of m/z 58 to m/z 41 is consistent with loss of ammonia by simple C–N bond cleavage.
In the main fragmentation pathway of 3a, the initial neutral loss of ammonia (m/z 144 → 127, Figures 2(a) and 2(b)) was followed by successive neutral losses of NO and CO (58 u) from 3b (m/z 127 → 97 → 69, Figure S4). The latter was also observed in the first step in the competing pathway of 3a (m/z 144 → 86, Scheme 1) and as the lowest energy fragmentation pathway of protonated 4-aminomethyl-3-hydroxyfurazan (1a).37 In these pathways, the development of a formal positive charge on N5 by protonation was the key determinant for ring cleavage. In the nucleophilic displacement of ammonia from 3a (Figure 4), alkylation generated a formal positive charge on N5, enabling N5–O bond cleavage and the subsequent loss of NO and CO.
The energy computed for the endergonic ring cleavage (3b(ol) → 3dp1ʹ, 180 kJ mol–1, Figure 5) was similar to that found for the nucleophilic displacement of ammonia (Figure 4). Subsequent low-energy conformational changes gave a suitable alignment for the exergonic abstraction of the proton on oxygen by nitrogen and the formation of a more stable iminium ion (3d). Homolytic bond cleavage with the release of NO and CO and formation of a cyclic radical cation (3e, m/z 69) had a modest barrier of 98 kJ mol–1. Cleavage of the ring in 3e, which was formed by the initial displacement of ammonia, led to the formation of a radical cation at m/z 41 (3f, HN + ≡C–CH2•, Figure S4A) and the loss of ethylene. The sequence of reactions for the fragmentation of 3a(ol) to 3f is shown in Scheme S1.
Potential energy profile for the fragmentation of 3b(ol) (m/z 127) showing ring opening and subsequent loss of NO and CO yielding the radical cation 3e (m/z 69).
Thus, an energetically feasible pathway for the fragmentation of 3a(ol) to 3e (m/z 69) was supported by the computations (Figures 4(a) and 5). However, a parallel pathway consisting of analogous fragmentation reactions of the higher energy lactam tautomer 3aʹ(one) is also mechanistically reasonable (Scheme S1). The energetic feasibility of the initial loss of ammonia from 3a(one) was shown by computations (Figure 4(b)) and, as outlined in Scheme S1, ring cleavage of 3b(one) followed by proton transfer generates 3d, the ion also formed by rearrangement of 3b(ol). Consequently, 3aʹ(one) is a possible, but higher energy, starting point for a second fragmentation pathway for the formation of product ion 3d (m/z 127) and the subsequent formation of the lower mass product ions 3e (m/z 69) and 3f (m/z 41).
Another possibility for the loss of ammonia is by an elimination mechanism. The abstraction of a proton from the propyl side chain by N5 in 3aʹʹ(ol) generates ion 3c(ol), a protonated allyl-substituted furazan (Figure S2A). Subsequent fragmentation of 3c(ol) by ring cleavage and losses of NO and CO is mechanistically possible (Scheme S2). As seen in the pathways initiated by nucleophilic displacement of ammonia (Figures 4 and 5; Scheme S1), a proton transfer step is needed before the loss of NO and CO occurs to form an acyclic radical cation (CH2 = CH–CH2–C•=NH2+) at m/z 69. A parallel sequence of analogous reactions for the fragmentation of 3aʹʹʹʹ(one) to the m/z 69 ion is also mechanistically feasible (Scheme S2). Homolytic C–C bond cleavage of the radical cation (m/z 69) produces an ion-neutral complex from which loss of ethylene requires hydrogen atom abstraction. If fragmentation of 3a proceeded by the routes outlined in Scheme S2, the formation of an ion at m/z 42 by dissociation of the ion-neutral complex and an ion at m/z 68 by loss of a hydrogen atom (see next section) is possible. However, neither ion was detected in the mass spectra of 3a (Figures 2 and S4A). The latter, along with the higher energy input computed for the initial elimination step (Figure S2A), suggest that this reaction sequence makes a smaller or negligible contribution to the fragmentation of 3a.
The elimination of ammonia by proton abstraction by the carbonyl oxygen in 3a(one) was studied computationally (Figure S3). Although a barrier of only 157 kJ mol–1 was located, the computations indicated that the elimination proceeded from 3aʹʹʹ(one). This higher energy conformation of the lactam tautomer is most likely a very minor component of the cations generated by ESI. Also, the energy required for the separation of ammonia from the product ion 3cp (m/z 127) was somewhat large (99 kJ mol–1). The proton in IN3c-NH3 is much closer to N than O and dissociation to the complementary3 ion NH4+ (m/z 18) and a neutral heterocycle may be favored. However, the ion 3cp produced by this elimination of ammonia is protonated at N2 of the ring. Unlike its protomer 3c(ol), neutral losses of NO and CO upon ring cleavage of 3cp is less likely and the formation of 3cp is therefore a poor fit with the experimental observations.
In principle, nucleophilic displacement of ammonia by the exocyclic oxygen is possible. In this instance, both the additional bond to oxygen and the location of the formal positive charge are incompatible with ring cleavage leading to the loss of both oxygen atoms in 3a(ol) and 3a(one) as NO and CO. As a result, the products formed by these mechanisms are a poor match with the experimental observations (Figures 2 and S4) and the reactions involving the exocyclic oxygen atom are unlikely to contribute to the loss of ammonia.
Overall, the analysis of secondary fragmentation processes and computed energetics indicate that the observed initial loss of ammonia occurs from 3a(ol) upon nucleophilic displacement by N5 in the ring.
Fragmentation pathway of 4a
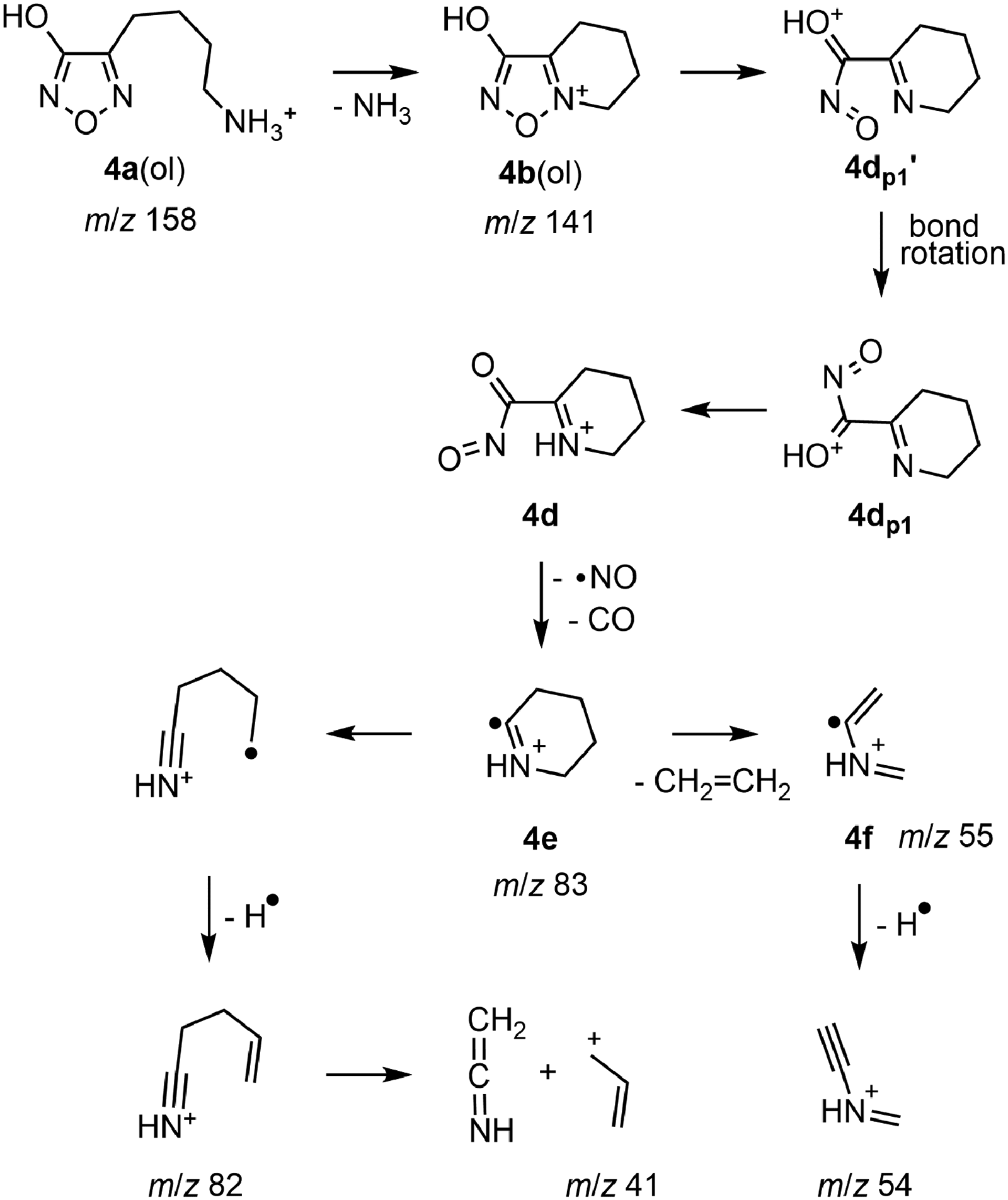
In accord with the structural relationship between protonated 4-(4-aminobutyl)-3-hydroxyfurazan (4a) and 3a (Figure 1), the tandem mass spectra collected for 4a (m/z 158; Figure S1) showed product ions at m/z 141, 83, and 55 that are homologs of ions observed in the mass spectra of 3a (Figures 2 and S4). The sequence of neutral losses (NH3, NO, CO and ethylene) observed for 4a was the same as that found in the main fragmentation pathway of 3a (Scheme S1), and the extension of the alkyl chain by one methylene unit had little or no effect on the fragmentation behavior of the homologous ion.
The similar relative energies computed for the various 4a and 3a ions (Table S3) suggested that the analogous 4a ions were likely starting points for the fragmentation pathways characterized for 3a (vide supra). Thus, fragmentation of 4a(ol) by the reaction sequence described for 3a (Figures 4(a) and 5; Scheme S1) led to a cyclic radical cation (4e) at m/z 83 (Scheme 2). Subsequent loss of ethylene gave the product ion at m/z 55 (4f). In the pseudo MS3 spectrum of m/z 141 (Figure S1, spectrum A inset) and at high collision energies (15–20 eV; Figure S1, spectra D and E), ions at m/z 82 and 54 accompanied the ions at m/z 83 and 55 indicating losses of a hydrogen atom. This loss of a neutral atom is consistent with the radical cation structures proposed for the ions at m/z 83 (4e) and 55 (4f).
A pathway for the fragmentation of protonated 4-(4-aminobutyl)-3-hydroxyfurazan (4a(ol)) showing the formation of the radical cation 4e by sequential neutral losses of NH3, NO and CO. Subsequent fragmentation of 4e occurred either by loss of CH2 = CH2 and a hydrogen atom or by the loss of a hydrogen atom and CH2 = C = NH.
As an alternative to the loss of ethylene from the cyclic radical cation 3e (Scheme S1), the neutral loss from the six-membered ring in 4e was attributed to ring cleavage by an electrocyclic process (Scheme 2). The analogous reaction has been proposed for the loss of ethylene from the 1-piperideine ion, a cyclic fragmentation product of LysH + .38
Conclusions
The predominant, initial fragmentation process observed for both the protonated 4-(3-aminopropyl)- and 4-(4-aminobutyl)-3-hydroxyfurazans 3a and 4a, respectively, was the neutral loss of ammonia. Computational analysis indicated that the process occurred by intramolecular, nucleophilic displacement of the protonated primary amine by N5 of the furazan ring. In previous studies,37 the ring protonated 4-(aminomethyl)- and 4-(2-aminoethyl)-3-hydroxyfurazans 1a and 2a, respectively, fragmented by ring cleavage with successive losses of NO and CO. Although the NO/CO neutral loss was also observed in the tandem mass spectra of 3a (Figure 2), it was observed only at higher collision energies. Thus, the homologous series of protonated heterocycles (1a–4a,Figure 1) fragmented by structure-specific processes depending on the protonation site and the length of the aminoalkyl side chain. By contrast, the anions formed by deprotonation of compounds 1–4 underwent ring cleavage upon CID with the formation of a common product ion (OCNO–).49
The computations indicated that fragmentation of the lactim tautomer 3a(ol) was the most energetically favorable process. Although similar barriers were found for the analogous reactions of lactam tautomer 3a(one), smaller amounts of this higher energy tautomer were likely formed by ESI.
The bicyclic product ion 3b(ol) formed by the loss of ammonia from 3a(ol) (Figure 4(a)) fragmented by 3-hydroxyfurazan ring cleavage with loss of NO and CO (Figure 5). In this cyclization process, a formal positive charge develops on N5 of the furazan ring and its fragmentation is analogous to the initial process characterized previously for 1a and 2a when protonated on N5.37
The present and previous37 computations indicated that the energy of the ion formed by protonation of the heterocyclic alkyl amine varied with protonation site, conformation, the extent of hydrogen bonding, and the tautomeric form of the heterocycle. The similar energetics provided several possibilities as starting points for fragmentation pathways and indicated that structural isomers of isobaric ions are generated during electrospray ionization. Nevertheless, the initial fragmentation behavior of the homologous heterocycles 1a–4a is significantly influenced by small structural changes such as the length of the alkyl chain and the site of protonation. The results further establish the importance of considering multiple variables for the interpretation/prediction of the fragmentation pathway of ions formed from multifunctional molecules.
Supplemental Material
sj-docx-1-ems-10.1177_14690667231214672 - Supplemental material for Intramolecular interactions and the neutral loss of ammonia from collisionally activated, protonated ω-aminoalkyl-3-hydroxyfurazans
Supplemental material, sj-docx-1-ems-10.1177_14690667231214672 for Intramolecular interactions and the neutral loss of ammonia from collisionally activated, protonated ω-aminoalkyl-3-hydroxyfurazans by J. Stuart Grossert, Donatella Boschi, Marco L. Lolli and Robert L. White in European Journal of Mass Spectrometry
Footnotes
Acknowledgements
Computational resources were generously provided by the Atlantic Computational Excellence Network (ACENET; https://www.ace-net.ca/) and Digital Research Alliance of Canada Compute Canada (). We thank X. Feng for maintenance of the mass spectrometers and for accurate mass determinations.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
Financial support was provided by the University of Turin (Grant LOLM_S1921_EX-POST_21_01; to M.L.L. and D.B.) and the Natural Sciences and Engineering Research Council of Canada (NSERC grant RGPIN/04536-2014; to R.L.W.).
ORCID iD
Robert L. White
Supplemental material
Supplemental material for this article is available online.
References
1.
JampilekJ. Heterocycles in medicinal chemistry. Molecules2019; 24: 3839.
2.
NiessenWMA. Fragmentation of toxicologically relevant drugs in negative-ion liquid chromatography–tandem mass spectrometry. Mass Spectrom Rev2012; 31: 626–665.
3.
NiessenWMA. Fragmentation of toxicologically relevant drugs in positive-ion liquid chromatography–tandem mass spectrometry. Mass Spectrom Rev2011; 30: 626–663.
4.
ShafieiMPeytonLHashemzadehM,et al.History of the development of antifungal azoles: a review on structures, SAR, and mechanism of action. Bioorg Chem2020; 104: 104240.
5.
KathiravanMKSalakeABChotheAS, et al.The biology and chemistry of antifungal agents: a review. Bioorg Med Chem2012; 20: 5678–5698.
6.
BuddeWL. Analytical mass spectrometry of herbicides. Mass Spectrom Rev2004; 23: 1–24.
7.
MeanwellNA. Applications of bioisosteres in the design of biologically active compounds. J Agric Food Chem2023 Articles ASAP.
8.
SainasSPippioneACBoschiD,et al.Hydroxyazoles as acid isosteres and their drug design applications–part 1: monocyclic systems. Adv Heterocyclic Chem2021; 134: 185–272.
9.
GomtsyanA. Heterocycles in drugs and drug discovery. Chem Heterocyclic Comp2012; 48: 7–10.
10.
BroughtonHBWatsonIA. Selection of heterocycles for drug design. J Mol Graphics & Modelling2004; 23: 51–58.
11.
PriyaHParanjothyM. Collision induced dissociation of deprotonated isoxazole and 3-methyl isoxazole via direct chemical dynamics simulations. J Am Soc Mass Spectrom2023; 34: 710–719.
12.
PengMLiSWuJ, et al.Fragmentation studies of sartans by electrospray ionization mass spectrometry. J Mass Spectrom2017; 52: 591–596.
13.
ZhangL-KPramanikBN. Characterization of major degradation products of an adenosine A2A receptor antagonist under stressed conditions by LC-MS and FT tandem MS analysis. J Mass Spectrom2010; 45: 146–156.
14.
MamerOALesimpleA. Protonated 1-methylimidazole decomposition by electrospray tandem mass spectrometry. Rapid Commun Mass Spectrom2005; 19: 1771–1774.
15.
ZhangJYXuFBreauAP. Collision-induced dissociation of valdecoxib metabolites: a novel rearrangement involving an isoxazole ring. J Mass Spectrom2004; 39: 295–302.
16.
ZhaoZWangQTsaiEW, et al.Identification of losartan degradates in stressed tablets by LC-MS and LC-MS/MS. J Pharm Biomed Anal1999; 20: 129–136.
17.
AdamsGWBowieJHHayesRN. Negative ion fragmentations of deprotonated heterocycles. The isothiazole, thiazole, isoxazole, and oxazole ring systems. Int J Mass Spectrom Ion Process1992; 114: 163–182.
18.
AdamsGWBowieJHHayesRN. Negative-ion fragmentations of deprotonated heterocycles. The pyrazole and imidazole ring systems. Rapid Commun Mass Spectrom1992; 6: 54–57.
19.
ChaiYChenHLiuX,et al.Formation of carbon dioxide attached fragment ions in the fragmentation of deprotonated tolfenpyrad and tebufenpyrad. J Am Soc Mass Spectrom2019; 30: 2060–2067.
20.
CaoXZhuKSongQ, et al.Proton-bound complex mediating retro-Michael-type fragmentation of protonated 3-substituted oxindoles in the Orbitrap high-energy collisional dissociation cell. Rapid Commun Mass Spectrom2015; 29: 515–520.
21.
CaoXZhangFZhuK, et al.Identifying the proton transfer reaction mechanism via a proton-bound dimeric intermediate for esomeprazoles by a kinetic method combined with density functional theory calculations. Rapid Commun Mass Spectrom2014; 28: 1045–1050.
22.
TuY-P. Dissociative protonation and fragmentation: retro-Friedel–Crafts reactions of heterocyclic drug and metabolite molecules in mass spectrometry. Int J Mass Spectrom2012; 316–318: 40–46.
23.
YouZGuoCPanY. An experimental and theoretical study on fragmentation of protonated N-(2-pyridinylmethyl)indole in electrospray ionization mass spectrometry. Rapid Commun Mass Spectrom2012; 26: 2509–2516.
24.
ZhangJChaiYJiangK, et al.Gas phase retro-Michael reaction resulting from dissociative protonation: fragmentation of protonated warfarin in mass spectrometry. J Mass Spectrom2012; 47: 1059–1064.
25.
PrakashCShafferCLNeddermanA. Analytical strategies for identifying drug metabolites. Mass Spectrom Rev2007; 26: 340–369.
26.
GillespieTAWingerBE. Mass spectrometry for small molecule pharmaceutical product development: a review. Mass Spectrom Rev2011; 30: 479–490.
27.
LolliMLHansenSLRolandoB, et al.Hydroxy-1,2,5-oxadiazolyl moiety as bioisoster of the carboxy function. Synthesis, ionization constants, and pharmacological characterization of γ-aminobutyric acid (GABA) related compounds. J Med Chem2006; 49: 4442–4446.
28.
LolliMLGiordanoCPickeringDS, et al.4-Hydroxy-1,2,5-oxadiazol-3-yl moiety as bioisoster of the carboxy function. Synthesis, ionization constants, and molecular pharmacological characterization at ionotropic glutamate receptors of compounds related to glutamate and its homologues. J Med Chem2010; 53: 4110–4118.
29.
MeanwellNA. Synopsis of some recent tactical application of bioisosteres in drug design. J Med Chem2011; 54: 2529–2591.
30.
LolliMLGiorgisMToscoP, et al.New inhibitors of dihydroorotate dehydrogenase (DHODH) based on the 4-hydroxy-1,2,5-oxadiazol-3-yl (hydroxyfurazanyl) scaffold. Eur J Med Chem2012; 49: 102–109.
31.
BallatoreCHurynDMSmithABIII. Carboxylic acid (bio)isosteres in drug design. ChemMedChem2013; 8: 385–395.
32.
PippioneACDosioFDucimeA, et al.Substituted 4-hydroxy-1,2,3-triazoles: synthesis, characterization and first drug design applications through bioisosteric modulation and scaffold hopping approaches. Med Chem Commun2015; 6: 1285–1292.
33.
PippioneACGiraudoABonanniD, et al.Hydroxytriazole derivatives as potent and selective aldo-keto reductase 1C3 (AKR1C3) inhibitors discovered by bioisosteric scaffold hopping approach. Eur J Med Chem2017; 139: 936–946.
34.
SainasSPippioneACGiorgisM, et al.Design, synthesis, biological evaluation and X-ray structural studies of potent human dihydroorotate dehydrogenase inhibitors based on hydroxylated azole scaffolds. Eur J Med Chem2017; 129: 287–302.
35.
LolliMLCarnovaleIMPippioneAC, et al.Bioisosteres of indomethacin as inhibitors of aldo-keto reductase 1C3. ACS Med Chem Lett2019; 10: 437–443.
36.
HorganCO’SullivanTP. Recent developments in the practical application of novel carboxylic acid bioisosteres. Curr Med Chem2022; 29: 2203–2234.
37.
GrossertJSBoschiDLolliML,et al.Fragmentation pathways arising from protonation at different sites in aminoalkyl-substituted 3-hydroxy-1,2,5-oxadiazoles (3-hydroxyfurazans). Rapid Commun Mass Spectrom2018; 32: 1403–1413.
38.
GrossertJSWhiteRL. Fragmentation reactions of protonated α,ω-diamino carboxylic acids: the importance of functional group interactions. J Mass Spectrom2021; 56: e4770.
39.
GrossertJSFancy PDWhiteRL. Fragmentation pathways of negative ions produced by electrospray ionization of acyclic dicarboxylic acids and derivatives. Can J Chem2005; 83: 1878–1890.
40.
FrischMJTrucksGWSchlegelHB, et al.Gaussian 09, Revision C.01, Gaussian, Inc., WallingfordCT, 2010.
41.
ChaiJ-DHead-GordonM. Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections. Phys Chem Chem Phys2008; 10: 6615–6620.
42.
WeinkamRJ. Reactions of protonated diamino acids in the gas phase. J Org Chem1978; 43: 2581–2586.
43.
WysockiVHBurinskyDJCooksRG. Competitive dehydration and deamination of α,ω-amino alcohols and α,ω-amino acids in the gas phase. J Org Chem1985; 50: 1287–1291.
44.
OgawaTZaitsuKKokajiT, et al.Development and application of a forensic toxicological library for identification of 56 natural toxic substances by liquid chromatography–quadrupole time-of-fight mass spectrometry. Forensic Toxicol2020; 38: 232–242.
45.
GinterováPSokolováBOndraP, et al.Determination of mushroom toxins ibotenic acid, muscimol and muscarine by capillary electrophoresis coupled with electrospray tandem mass spectrometry. Talanta2014; 125: 242–247.
46.
GonmoriKHasegawaKFujitaH, et al.Analysis of ibotenic acid and muscimol in Amanita mushrooms by hydrophilic interaction liquid chromatography–tandem mass spectrometry. Forensic Toxicol2012; 30: 168–172.
47.
GibbonsJPughJDimopoulos-ItalianoG,et al.A qualitative study of amlodipine and its related compounds by electrospray ionization tandem mass spectrometry. Rapid Commun Mass Spectrom2006; 20: 1715–1723.
48.
YasudaTTanakaMIbaK. Quantitative determination of amlodipine in serum by liquid chromatography with atmospheric pressure chemical ionization tandem mass spectrometry. J Mass Spectrom1996; 31: 879–884.
49.
GrossertJSPippioneACBoschiD, et al.Heterocyclic ring cleavage upon collision-induced dissociation of deprotonated 3-hydroxy-1,2,5-oxadiazoles (3-hydroxyfurazans). J Mass Spectrom2015; 50: 1433–1437.
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.