
Abstract
The elemental impurities in pharmaceutical products have aroused widespread concern among respective supervising authorities and official pharmacopoeias since they are harmful and have no therapeutic effects. Metronidazole benzoate is used extensively to treat a variety of infections. However, impurities will inevitably be introduced in the manufacturing process of metronidazole benzoate. Hence, in this study, a sensitive method was developed for trace determination of elemental impurities in metronidazole benzoate active pharmaceutical ingredients by using inductively coupled plasma mass spectrometry in kinetic energy discrimination mode. The method was validated for system suitability, specificity, linearity, sensitivity, accuracy, and precision according to USP chapter <233> Elemental Impurities-Procedure. The method had good linearity with correlation coefficients > 0.99. The limits of detection were in the range of 0.0003–0.1411 μg/g, which was lower than the acceptable limit and indicated the high sensitivity of the method. The method was accurate with the recoveries in the range of 92%–107%. Moreover, the content of seven elemental impurities in the three batches of metronidazole benzoate active pharmaceutical ingredients by this method was originally below their limits and less than 30% of permitted daily exposure, meeting the requirement of International Council for Harmonization Q3D guidelines. Thus, this newly developed and validated method for estimating elemental impurities in metronidazole benzoate active pharmaceutical ingredients was within the permitted limit and suitable for routine use.
Keywords
Introduction
Metronidazole benzoate (MB), the prodrug of metronidazole, is extensively used to treat a variety of infections, including amoebic dysentery, trichomonasis, periodontitis, giardiasis, and other anaerobic infections, by hydrolyzing and releasing metronidazole after oral administration in the gastrointestinal tract.1,2 However, impurities, for example, organic impurities (process and drug-related), inorganic impurities, and residual solvents can be produced in the manufacturing process of MB active pharmaceutical ingredients (APIs), which makes it risky in the process of use. Hence, it is of great significance to control these potential impurities in MB APIs.
Impurity control is a crucial step in maintaining the high quality and safety of pharmaceutical products. Recently, the elemental impurities (EIs) in pharmaceutical products have aroused widespread concern among respective supervising authorities and official pharmacopoeias. The reason behind this concern lies in the inherent toxicity of these impurities and their ability to catalyze the decomposition of APIs, ultimately presenting hazards to human health.3,4 A new paradigm for the control and risk assessment of EIs in pharmaceuticals has been established with the publication of the Guideline for Elemental Impurities (Q3D) by the International Council for Harmonization (ICH). The guideline sets permitted daily exposure (PDE) in μg/day for 24 elements from three routes of administration (oral, parenteral, and inhalation). The elements are grouped into four classes based on their toxicity and the likelihood of occurrence in pharmaceutical products: Class 1 (As, Cd, Hg, and Pb), Class 2A (Co, Ni, and V), Class 2B (Ag, Au, Ir, Os, Pd, Pt, Rh, Ru, Se, and Tl), Class 3 (Ba, Cr, Cu, Li, Mo, Sb, and Sn). According to ICH Q3D guideline, when considering the risk of oral drugs, elements classified as Class 2B and Class 3 can be excluded from the risk assessment unless they are intentionally added. Furthermore, the guideline introduced the important concept of control threshold, which is defined as 30% of the PDE. If the measured value of the introduced EIs from all sources is less than the control threshold, there is no need for additional control of the EIs.5–7
With the implementation of ICH Q3D guideline, the method of controlling EIs has changed from traditional “heavy metal testing” to science-based risk assessment. Therefore, conventional methods such as the colorimetric method and sulfide precipitation method, which don’t meet the criteria for specific detection of trace and ultra-trace levels of EIs in APIs and other pharmaceutical products, have replaced by advanced analytical methods like atomic absorption spectroscopy, X-ray fluorescence spectrometry, instrumental neutron activation analysis, inductively coupled plasma-optical emission spectrometry (ICP-OES), and inductively coupled plasma mass spectrometry (ICP-MS).8,9 ICP-based methods show advantages of low limit of detection (LOD) (μg/L for ICP-OES and ng/L for ICP-MS), multi-element simultaneous analysis, wide linear range, and isotopic analysis. They are recommended for the determination of EIs by USP chapter <233>.10–13 By comparison, the ICP-MS method with better sensitivity and specificity is widely applied in ultra-trace analysis.5,9 Recently, a new type of mass spectrometry called triple quadrupole ICP-MS (ICP-QQQ) has emerged. Compared with single quadrupole mass spectrometry, ICP-QQQ greatly improves the mass filtering power of the instrument by introducing extra two quadrupoles. 14 Thus, it can overcome complex polyatomic interferences and provide lower detection limits and background signal 15
Since there is no method for detecting EIs in MB APIs, an ICP-MS method with kinetic energy discrimination (KED) mode was developed for trace determination of 7 EIs in MB APIs, including Cd, Pb, As, Hg, Co, Ni, and V, which require risk assessment for oral administration. Furthermore, the method was validated in terms of system suitability, specificity, linearity, LOD, limit of quantification (LOQ), accuracy, and precision according to the requirements given in USP chapter <233> Elemental Impurities-Procedure.
Experimental
Reagents and standards
Ultrapure water used in the experiment was prepared by passing water from a Cascada III ultra-pure water system (Pall, USA). Ultra-pure grade nitric acid (Suzhou Jingrui Chemical Co., Ltd (China)) was used for sample preparation. Standard Pb (100 mg/L), standard Hg (100 mg/L), standard Co (100 mg/L), standard V (100 mg/L), standard Ni (100 mg/L), standard Au (1000 mg/L), standard Bi (1000 mg/L), standard Te (1000 mg/L) were purchased from Inorganic Ventures (USA). Standard Cd (100 mg/L), standard As, and standard Ge (1000 mg/L) were obtained from the National Center of Analysis and Testing for Nonferrous Metals and Electronic Materials (China). MB APIs for testing and validation were supplied by Hubei Hongyuan Technology Co., Ltd (Hubei, China).
Sample solutions
0.2 g of MB APIs was weighed, and 0.1 mL of 100 μg/mL standard gold solution, 5 mL of HNO3 were added in a tetrafluoroethylene beaker. After dissolving, the solution was transferred to a 50 mL volumetric bottle. And the sample solution was prepared by diluting to the scale using acid mixture (HNO3:HCl = 8:1, v/v).
The sample blank solution was prepared using the same method above without adding MB APIs. And it was used for LOD and LOQ studies.
Spiked samples were prepared at 30% (0.3 J), 100% (1.0 J), and 150% (1.5 J) of the target limit using the same method of sample solution (J is the limit value of concentration). The difference was that 0.15, 0.5, and 0.75 mL of standard stock solution was added at the same time as MB APIs. Six preparations at the 100% spiking level were prepared, and three preparations at 30% and 150% spiking level were prepared.
Instrumentation
All element determinations were performed on an ICP-MS system (NexION1000, Perkin Elmer, USA) equipped with standard nickel sampling and skimmer cones, concentric nebulizer, cyclonic spray chamber, and collision cell operating in KED mode to overcome spectral interferences. The instrument with all matrix solutions and extended dynamic range features enables the analysis of samples with high solid dissolved content and organic samples by using argon dilution and organic oxygenation. Argon (99.999%) was used for plasma generation and worked as a nebulization and auxiliary gas. Helium with high purity (99.999%) was used as a collision gas. The radio frequency power of 1600 W, the nebulizer flow rate of 0.96 L·min−1, the collision gas rate of 4.0 mL·min−1, the plasma gas flow rate of 15.0 L·min−1, the auxiliary gas flow rate of 1.2 L·min−1, and the peristaltic pump speed of 35 r·min−1 were used.
The most abundant isotopes such as 111Cd, 208Pb, 75As, 202Hg, 59Co, 51V, and 60Ni were determined, while 74Ge, 130Te, and 209Bi were selected as the internal standard (IS) element. The determination of isotopes in MB APIs solutions was carried out using ICP-MS with KED mode. All samples were injected manually. The instruments were turned systematically. Daily performance testing (check instrument performance, nebulizer gas flow optimization, sensitivity, CeO/Ce, etc.) was conducted before batch analysis to achieve the best experimental conditions.
Method validation
The method for quantification of the 7 EIs in MB APIs using ICP-MS with KED mode was validated for system suitability, specificity, linearity, LOD, LOQ, repeatability, intermediate precision, and accuracy according to USP chapter <233>. Details are described in the “Results and Discussion” section.
Results and discussion
Development of ICP-MS method
ICP-MS and ICP-OES were recommended in the USP chapter <233> owing to the multi-element analysis and relatively low LOD. 10 Compared with ICP-OES, ICP-MS with higher sensitivity was selected in this study. Considering that MB can be dissolved in nitric acid, and direct dissolution in acid is simple, fast, and practical. Thus, direct dissolution was chosen to prepare sample solution. The high volatility and memory effect of mercury lead to incomplete recovery.16,17 Therefore, HCl was added to the diluent to stabilize mercury in the solution, and gold was selected as the stabilizer to prevent the volatilization and loss of mercury. There are usually spectroscopic interference and non-spectroscopic interference in ICP-MS testing. In order to eliminate the spectral interference of ICP-MS, helium was used as the collision gas, and KED mode was chosen in this study. Then, in KED mode, based on the principle of high isotope abundance and low interference factor, 111Cd, 208Pb, 75As, 202Hg, 59Co, 51V, and 60Ni were used as the analysis objects. Internal standardization can effectively correct the interference from the matrix effect and the changes in instrument operating conditions. Ideal IS should have the characteristic of similar mass and ionization potential to the analyte, and absence in sample. 11 Therefore, 74Ge was chosen as the IS for 51V, 59Co, 60Ni, and 75As, 130Te was chosen as the IS for 111Cd, and 209Bi was chosen as the IS for 208Pb and 202Hg.
Method validation
The method validation was performed according to USP chapter <233>.
System suitability
The system suitability of the method was evaluated by injecting 100% of the limit concentration solution before and after the determination of the sample solution into ICP-MS system, and calculating the signal drift. The mean signal drifts of As, Cd, Pb, V, Co, Ni, and Hg were 4%, 1%, 1%, 0%, 1%, 1%, and 1%, respectively, which were in accordance with the requirements in USP chapter <233> (less than 20%), indicating that the ICP-MS system was suitable to detect EIs in MB APIs.
Specificity
The specificity of the analytical method was evaluated by injecting calibration blank solution, sample blank solution, sample solution, spiked sample solution, and standard solution to the analytical system. The results showed that the signal ratio of calibration blank/standard solution was <2%, and the signal ratio of sample blank/standard solution was <3%. Each element has its own m/z and there were no other impurities signal and interference found in each individual element to be measured, which indicated the method had good specificity.
Linearity and range
The linearity of EIs in MB APIs was analyzed by injecting calibration standards with the concentration of the target element in the range of 0%–250% of the limit concentration. The calibration curve was constructed with the intensity ratio of EI/IS as the ordinate and the concentration as the abscissa. The standard calibration curves showed good linearity with correlation coefficient (R) for each analyst >0.99 over the proposed concentration. The regression equations, calibration range, and correlation coefficients of each element are shown in Table 1.
Validation results of the studied impurities in MB APIs.
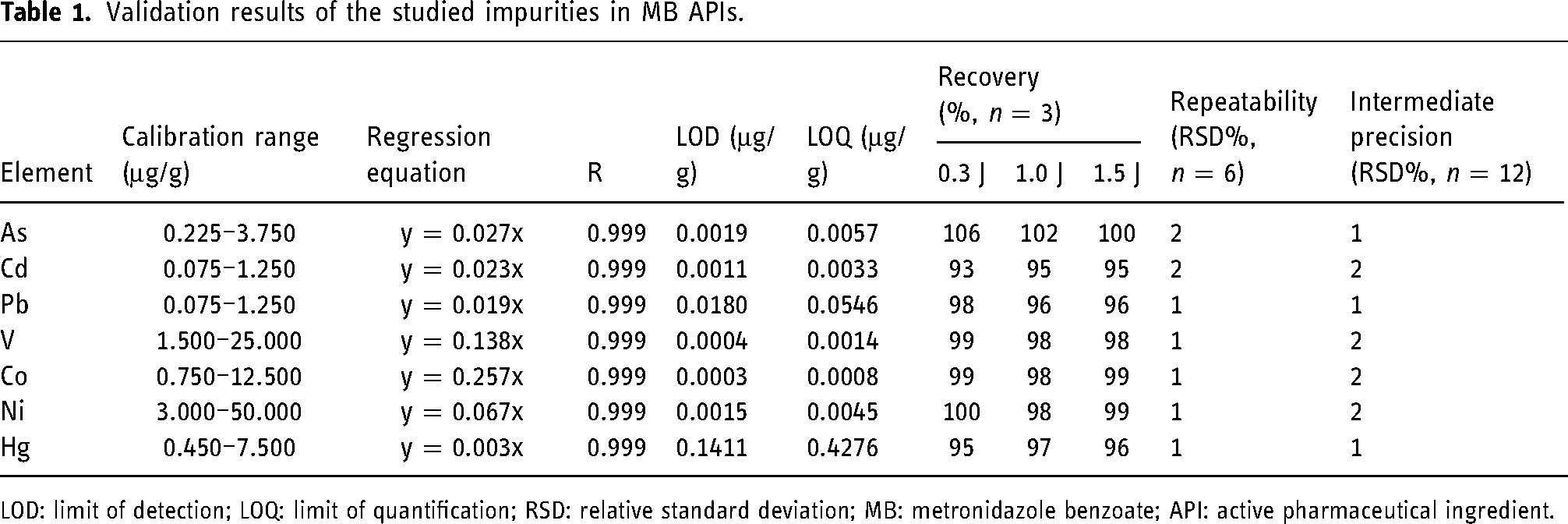
LOD: limit of detection; LOQ: limit of quantification; RSD: relative standard deviation; MB: metronidazole benzoate; API: active pharmaceutical ingredient.
LOD and LOQ
LOD and LOQ of the method were performed using the standard deviation of the blank intensity (δ) and the slope of the calibration curve (S). The LOD can be determined as the analyte concentration equal to 3.3δ/S. LOQ can be defined as the analyte concentration equal to 10δ/S. The LOQs were in the range of 0.0008–0.4276 μg/g, providing details about the least amount that the method can calculate accurately. LODs were in the range of 0.0003–0.1411 μg/g, which was lower than the acceptable limits in MB APIs and indicated the high sensitivity of the method. The LODs and LOQs for each element are shown in Table 1.
Repeatability
The repeatability of the method was performed by analyzing six 100% target limit spiked samples and evaluating the degree of repeatability among the replicate results. The relative standard deviations (RSDs) of As, Cd, Pb, V, Co, Ni, and Hg were 2%, 2%, 1%, 1%, 1%, 1%, and 1%, respectively (Table 1, Table S3), which were significantly less than 20% required in USP chapter <233>, indicating that the method showed good precision under repeatability conditions.
Intermediate precision
The same six 100% target limit spiked solutions were freshly prepared and analyzed by another analyst on a different day. The samples were quantified against fresh calibration standards. The RSDs of the newly prepared six independent replicates by this analyst for As, Cd, Pb, V, Co, Ni, and Hg were 1%, 3%, 2%, 2%, 2%, 2%, and 2%, respectively (Table S4). Intermediate precision was established as operator/day to operator/day precision change in the mean. The overall RSDs of two analysts for As, Cd, Pb, V, Co, Ni, and Hg were 1%, 2%, 1%, 2%, 2%, 2%, and 1%, respectively (Table 1, Table S4). The method exhibited good intermediate precision for RSDs < 25%.
Accuracy
The accuracy of the method was analyzed by injecting 30% spiked solution (0.3 J), 100% spiked solution (1.0 J), and 150% spiked solution (1.5 J) into ICP-MS system and calculating the spiked recoveries of target elements. From the obtained results in Table 1 and Table S5, it was known that the spiked recoveries for the mean of three replicate preparations at each concentration were in the range of 70%–150%, in accordant to USP chapter <233>, indicating that the method was accurate.
Sample test
According to this method, EIs of three standard production batches of MB APIs were measured, and the results were presented in Table 2. Results showed that the content of EIs in three batches of MB APIs was far less than the control threshold (30% PDE), indicating that additional controls were not required.
Results of sample test.
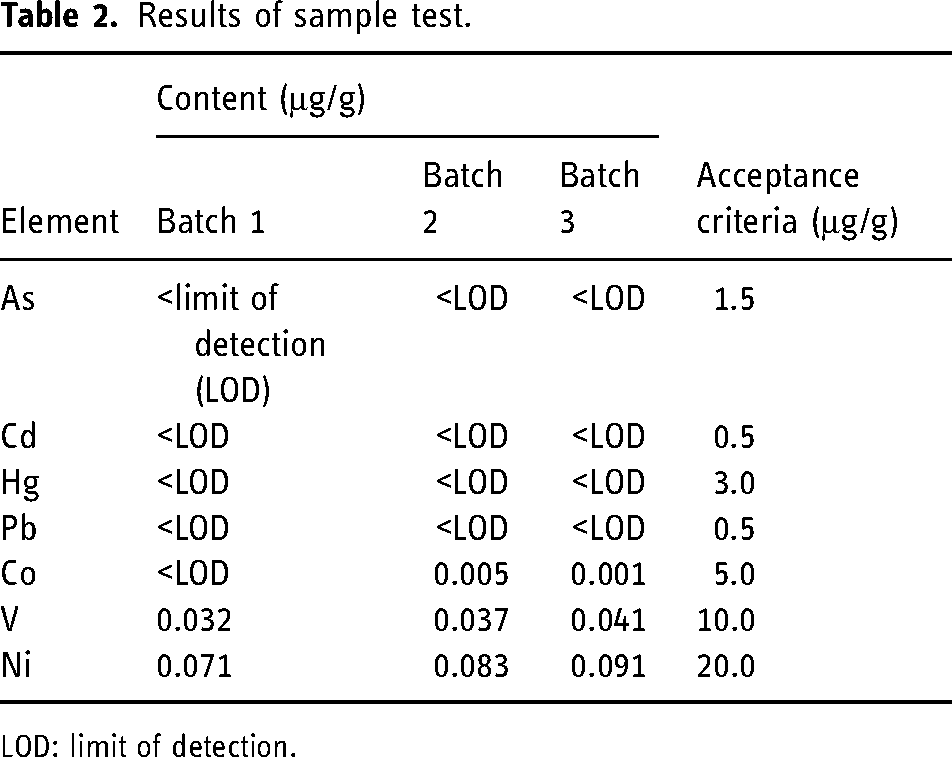
LOD: limit of detection.
Conclusion
In this paper, an ICP-MS method for the determination of seven EIs in MB APIs was developed and validated for system suitability, specificity, linearity, LOD, LOQ, repeatability, intermediate precision, and accuracy according to USP chapter <233>. The method was sensitive and specific with good accuracy and precision for the determination of EIs in MB APIs. Seven EIs in 3 batches of MB APIs were determined, and the contents were much lower than 30% of PDE. The target EIs in MB APIs were evidenced risk-free and no further control was required. This study provided a reference for the risk assessment of EIs in MB APIs in the pharmaceutical industry. To be improved, sample preparation in our work is not environmentally friendly, and a green sample preparation method should be considered in future research.
Supplemental Material
sj-docx-1-ems-10.1177_14690667231211696 - Supplemental material for Method for determination of elemental impurities in metronidazole benzoate using inductively coupled plasma mass spectrometry
Supplemental material, sj-docx-1-ems-10.1177_14690667231211696 for Method for determination of elemental impurities in metronidazole benzoate using inductively coupled plasma mass spectrometry by Maoxian Tian, Hui Zhang, Huajun Fan, Mingxing Yin, Wenqing Wang and Chunyang Shi in European Journal of Mass Spectrometry
Footnotes
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.