
Abstract
Kinetic rate constants for the oxidation reaction of the hydroxyl radical with CH3SH, C2H5SH, n-C3H7SH, and iso-C3H7SH under inert (Ar) conditions over the temperature range 252–430 K have been studied theoretically using density functional theory along with various exchange–correlation functionals as well as the benchmark CBS-QB3 quantum chemical approach.
Bimolecular rate constants were estimated using transition state theory and the statistical Rice–Ramsperger–Kassel–Marcus theory. Comparison with experiment confirms that in the OH addition reaction pathways leading to the related products, the first bimolecular reaction steps have effective negative activation energy barriers. Effective rate constants have been calculated according to a steady-state analysis of a two-step model reaction mechanism. As a consequence of the negative activation energies, pressures higher than 104 bar are required to reach the high-pressure limit. Both from thermodynamic and kinetic viewpoints, the most favorable process here is the oxidation reaction of hydroxyl radicals with n-C3H7SH.
Keywords
Introduction
The emission of gaseous sulfur compounds into the atmosphere is due to both natural and anthropogenic processes. 1 In recent years, investigation of the global atmospheric sulfur cycle has been the subject of intense scientific interest because of the need to assess the contribution of anthropogenically produced sulfur to problems such as acid rain, visibility reduction, and climate modification. 2 In heavily industrialized regions, anthropogenic sulfur emissions exceed natural emissions by about an order of magnitude. However, on a global scale, natural sulfur emissions are thought to equal approximately those from anthropogenic sources.3,4 Despite their low concentrations, larger organic sulfur compounds may play an important role in tropospheric sulfur chemistry. All hydrogen-containing sulfur compounds (such as R-SH, R: CH3, C2H5, n-C3H7, iso-C3H7) are probably removed from the atmosphere very rapidly by reaction with OH radicals, and some may also react rapidly with NO3. 2
The hydroxyl radical is a reactive intermediate of importance in combustion processes and in atmospheric chemistry. 5 Hence, the kinetic rate constants and mechanisms for the reaction of hydroxyl radicals with organic sulfides (R-SH), which play a dominant role in the troposphere,6–15 are needed in order to understand their involvement in photochemical air pollution. The products of the [R-SH···OH]· reaction chain have not been established, and the laboratory data can be interpreted either as OH addition or as H-abstraction reaction pathways. 1 Since there are no reported theoretical data for these reactions, in this article, absolute kinetic rate constants for the reactions of OH radicals with the studied compounds have been determined over the temperature range of 252–430 K.
The reaction of OH radicals with CH3SH was studied experimentally by Wine et al. 2 and Atkinson et al. 5 over the temperature range of 299–426 K with typically 50 Torr of argon as the diluent gas by using the flash photolysis-resonance fluorescence technique. The Arrhenius expression obtained was k(MeSH) = 8.89 × 10−12 exp((790 ± 300)/RT) cm3 molecule−1 s−1 with a rate constant at 298 K of (3.39 ± 0.34) × 10−11 cm3 molecule−1 s−1. Analogous increases in kinetic rate constants have previously been observed for the reactions of O (3P) atoms with CH3SH and C2H5SH at room temperature.16,17 The observation of a negative Arrhenius activation energy for the reaction of OH radicals with CH3SH over the studied temperature ranges probably arises from a zero or near-zero activation energy combined with a temperature-dependent preexponential factor, as has been previously observed and discussed for the reactions of OH radicals with olefins.18–20 CH3SH is thus as reactive as olefins toward OH radicals and can be calculated to have a half-life due to reaction with OH radicals in the lower troposphere, using an OH radical concentration of ∼3 × 106 molecule cm−3, of ∼2 h.21–25
The discharge flow experiments of Mac Leod et al. 25 and Lee and Tang, 26 which were carried out at total pressures 10−100 times lower than those employed in the flash photolysis studies, report kinetic rate constants for OH•+CH3SH and OH•+C2H5SH, which are slightly lower than those obtained in the flash photolysis studies. On the other hand, Cox and Sheppard, 27 using a relative rate technique, obtained a 298 K rate constant for OH•+CH3SH in 1 atm air that is nearly a factor of three higher than that obtained in the flash photolysis studies. Cox and Sheppard suggested that the difference between their result and the flash photolysis results could be explained by assuming that the OH•+CH3SH rate constant was pressure-dependent. While the flash photolysis results agree with the discharge flow results within the combined experimental errors, the consistently lower rate constants obtained in the discharge flow studies suggest that OH reactions with CH3SH and C2H5SH may be pressure-dependent in the low-pressure regime. 2
There are three possible pathways for the reactions of OH with thiols:
Abstraction of hydrogen from a carbon atom [CH3SH+OH•→ H2O+•CH2SH].
Abstraction of hydrogen from the sulfur atom [CH3SH+OH•→ H2O+CH3 S•].
Addition to the sulfur atom [CH3SH+OH•→ CH3SH-OH•].
Observations show that the kinetic rate constant related to the first pathway at room temperature is nearly independent of the nature of the hydrocarbon chain, which clearly indicates that hydrogen abstraction from a carbon atom (C−H bond) is a negligible reaction channel for all reactions investigated. 2 Thus, these reactions must proceed via either H-abstraction from the weak S−H bonds (of bond dissociation energy 91 ± 1.5 kcal mol−1)28,29 or by the formation of an OH–thiol adduct (see Scheme 1). Since no definitive information is available concerning this issue, further experimental data are required concerning the dynamics of the reactions of the initial OH radicals with the aliphatic thiols containing short-chain alkyl groups (1 to 3 carbon atoms) and of the subsequent reaction pathways operative under atmospheric conditions.
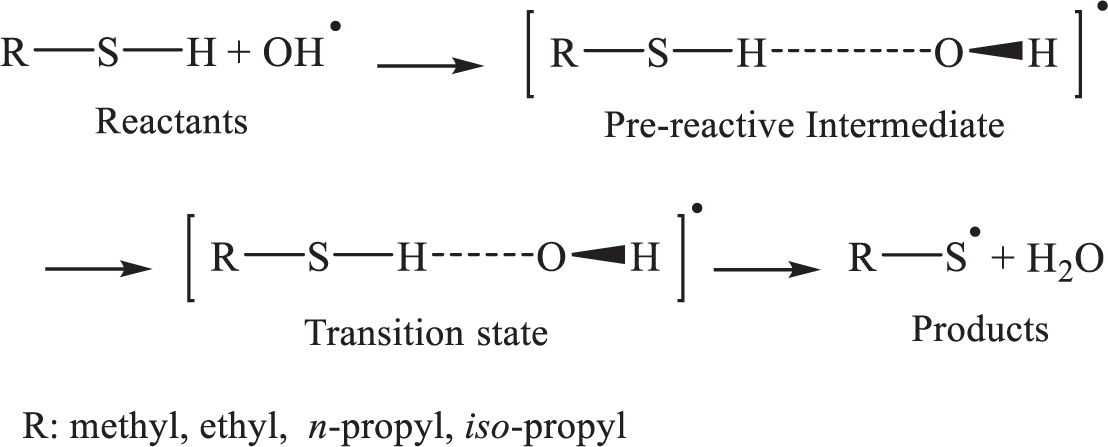
H-abstraction reaction pathways of aliphatic thiols containing alkyl groups (1 to 3 carbon atoms) by hydroxyl radicals yielding water and the related thiol radicals.
The reactions of hydroxyl radicals with the aliphatic thiols such as methanethiol (

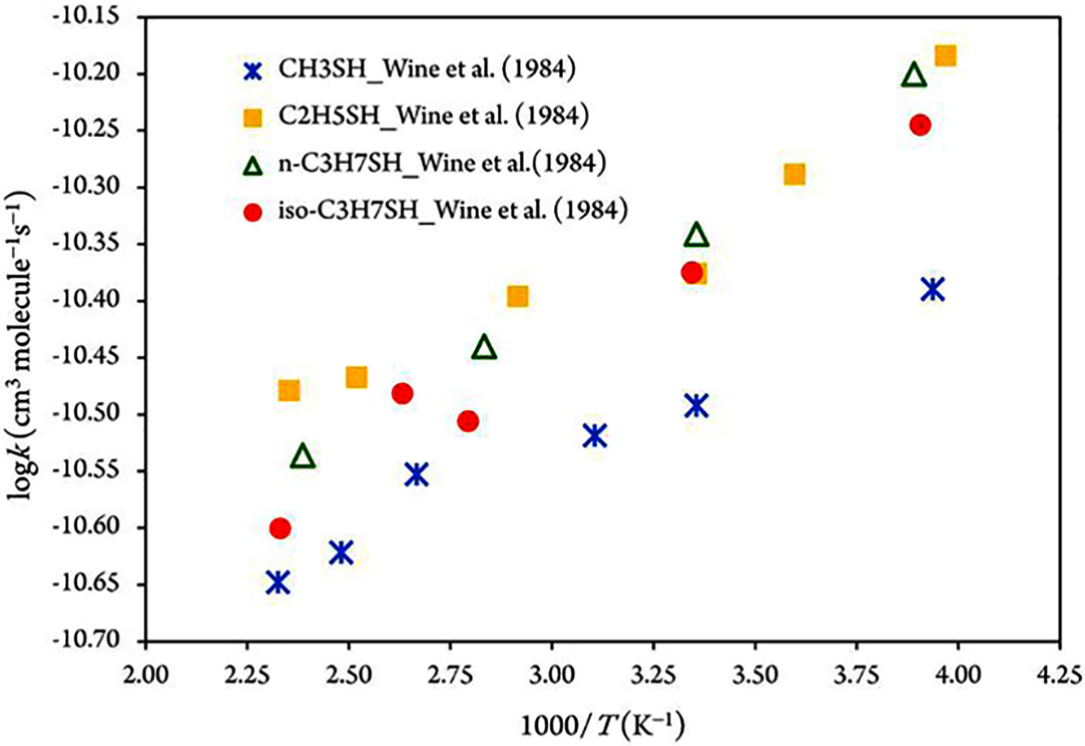
Arrhenius plots of the rate constants for the reactions of OH radicals with compounds
The Arrhenius rate constants decrease gradually with increasing temperature at temperatures ranging from 252 to 430 K, confirming negative activation energies.
To explain the disparity of the kinetic data shown in Figure 1, pathways
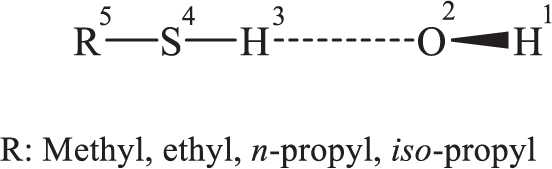
Retained atom labeling for characterizing the structures of intermediates and transition states.
To seek more quantitative insights into these reaction mechanisms, comparison will be made with benchmark theoretical calculations employing the high-level composite CBS-QB3 ab initio approach,30–38 to determine which exchange–correlation functional gives the most accurate energy barriers and reaction energies. We note that density functional theory (DFT) methods alone were found to be insufficient for quantitatively investigating the potential energy surfaces of the reaction mechanisms and kinetics of oxidation processes of benzene 39 and naphthalene40,41 by OH radicals, given the inability of many popular DFT functionals to describe quantitatively nonbonded interactions and barrier heights.
The aim of the present study was to achieve comprehensive and quantitative theoretical insights of the studied pathways. For this purpose, we first used DFT along with the dispersion-corrected ωB97XD 42 and the M06-2x 43 exchange–correlation functionals and the aug-cc-pVTZ basis set 44 and then compared the reaction energies and energy barriers obtained with the results of benchmark theoretical calculations employing the CBS-QB3 composite method. In addition, kinetic rate constants in the high-pressure limit were obtained using transition state theory (TST),45–51 and their falloff behavior was investigated at lower pressures using the statistical Rice–Ramsperger–Kassel–Marcus (RRKM) theory52–54 to unravel the available experimental data2,5 at temperatures ranging from 252 to 430 K.
Theory and computational details
All calculations reported in this study were performed using the Gaussian09 package of programs. 55 Molecular structures were visualized with GaussView. 56 The geometries and harmonic vibrational frequencies of all stationary points involved in the reactions of interest were determined using DFT 57 in conjunction with the ωB97XD and M06-2x functionals and the aug-cc-pVTZ basis set.
The energies of all the studied stationary points were reevaluated using the CBS-QB3 model.30–38 This model includes low-level calculations using large basis sets, mid-sized basis sets for second-order correlation corrections, and small basis sets for high-level correlation corrections. It also includes an energy extrapolation up to the coupled cluster theory including single, double, and perturbative triple excitations (CCSD(T)) level58−60 in complete basis set limits in order to correct second-order Møller–Plesset correlation energies. 59
The five-step CBS-QB3 series of calculations starts with a geometry optimization at the B3LYP theoretical level,33,38,61 followed by calculation of vibrational frequencies and thermodynamic state functions 62 obtained from canonical partition functions that were computed for an ideal gas using Boltzmann statistical thermodynamics.63−66 A weakness of the B3LYP approach is that it neglects dispersion forces which may have some influence on torsional characteristics. The CBS-QB3 method amounts to an extrapolation of energies to the CCSD(T) level in conjunction with a complete basis set. The CBS-QB3 method was developed with the idea that a major source of error in quantum mechanical calculations arises from truncation of the basis set.31,33,35,37 The CBS-QB3 method is a benchmark quantum chemical approach in order to calibrate the accuracy of DFT methods employing necessarily approximate exchange–correlation functionals.
The intrinsic reaction coordinate (IRC) analysis67,68 was carried out in both directions (forward and backward) along the reaction path using the Hessian-based predictor corrector integrator algorithm in order to check the energy profiles connecting the identified transition state (TS) structures to the associated energy minima.69,70
Analysis of the oxidation of aliphatic thiols by OH radicals turned out to be consistent with the scheme, 71 which assumed that this reaction occurs according to a two-step mechanism, 72 involving first a fast preequilibrium between the reactants (R−SH+OH•) and a prereactive complex [R−SH···OH]• (IM i ), followed by H-abstraction leading to the related products
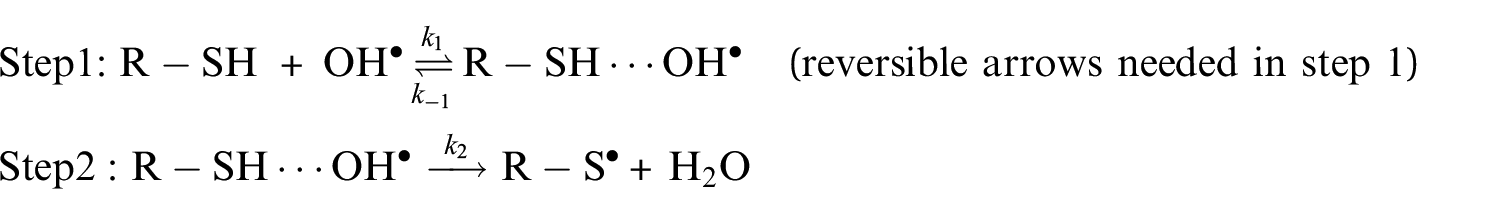
where k1 and k−1 denote the rate constants for the forward and reverse reactions associated with the first step, and k2 is the rate constant corresponding to the second step. The overall rate constant can be rewritten as

where Kc = k1/k−1 is the equilibrium constant for fast preequilibrium between the reactants and the prereactive complex (see Table S1 of the Supplemental Material). In the high-pressure limit (TST), the kinetic rate constant characterizing the unimolecular dissociation reaction is given by45−52,73
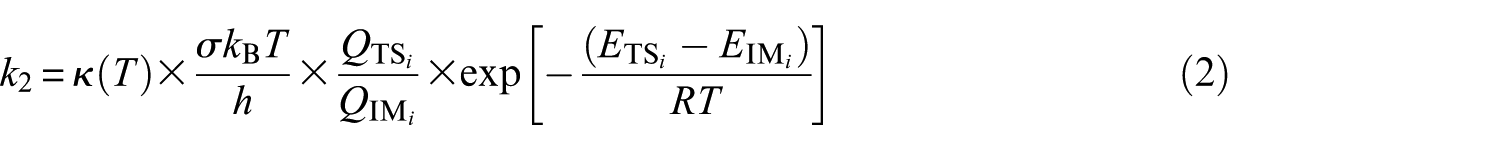
where R denotes the ideal gas constant, and kB and h represent Boltzmann’s and Planck’s constants, respectively. In the equation, QR−SH, QOH, QIMi, and QTS i represent the total molecular partition functions for the isolated reactants, prereactive molecular complex (IM i ), and transition state (TS i ) associated with the unimolecular dissociation reaction (step 2), respectively. ER−SH, EOH, EIMi, and ETSi are the corresponding energies including B3LYP/6-311G(2d,d,p) estimates for zero-point vibrational contributions. In the above equation, σ is the reaction-path degeneracy and κ(T) denotes Wigner’s74,75 tunneling correction factor (see Table S2 of the Supplemental Material)

where Im(υ1) is the imaginary vibrational frequency of the relevant TS, and R is the ideal gas constant. Kinetic rate constants for the channels studied were calculated over the temperature range of 252–430 K and in the high-pressure limit (1 atm) using TST. Unimolecular kinetics are given by76,77
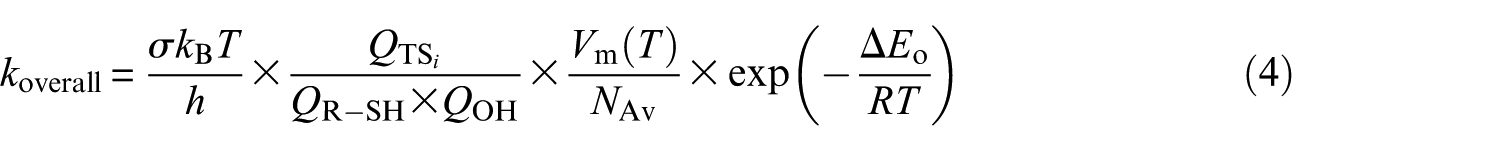
Kinetics for each pathway were evaluated in the high-pressure limit and using B3LYP/6-311G(2d,d,p) molecular partition functions that were computed using the vibrational ground state as energy reference along with the CBS-QB3 estimates for activation energies. Due to the simplicity of conventional TST, it gives the upper limit of the kinetics and enables us to provide reliable rate constants in the limiting high-pressure behavior.54,78
According to the RRKM theory, the energy-dependent microcanonical rate constants, k(E), for the unimolecular decomposition of a molecule with internal energy E can be expressed as 79

where G†(E − Eo) is the sum of the densities of states of the TSs at energies ranging from 0 to E − Eo, where Eo is the critical energy for the reaction, and N(E) is the density of vibrational states in the activated molecule at an energy E. G†(E − Eo) and N(E) were computed using the Beyer–Swinehart exact counting algorithm 79 improved by Stein and Rabinovitch. 80 Canonical (temperature-dependent) RRKM rate constants were determined from state integration and Boltzmann averaging.
TST and RRKM kinetic rate constants for the reaction pathways
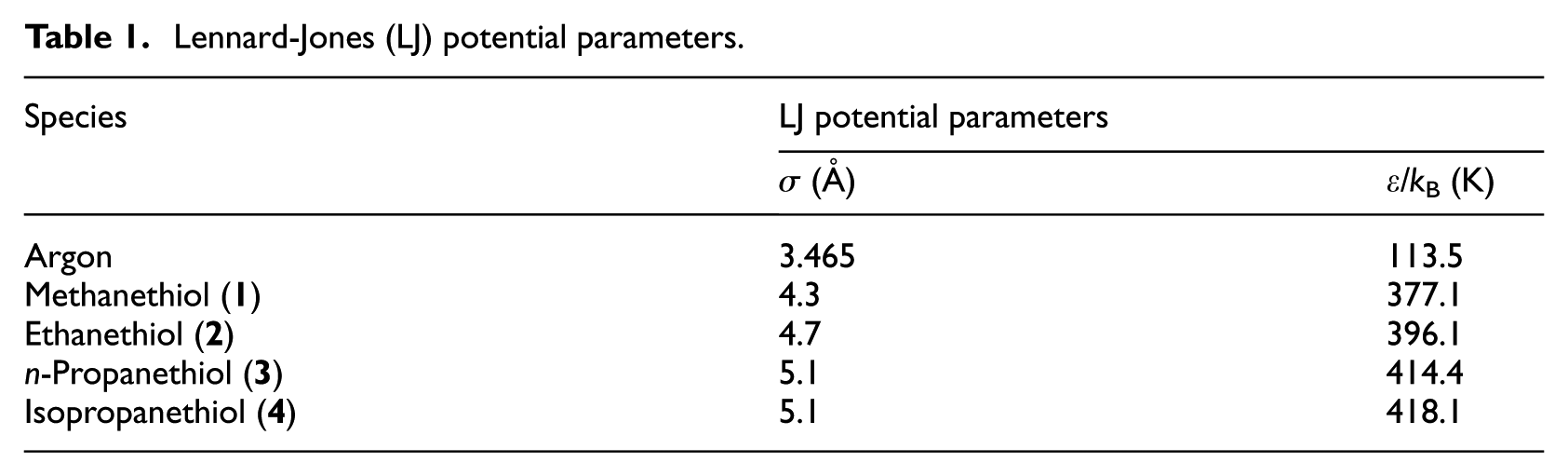
Lennard-Jones (LJ) potential parameters.
Results and discussion
Structural characteristics of stationary points
The optimized molecular structures of the intermediate complexes, TSs, and products along reaction pathways
Structural parameters for all stationary points involved in the H-abstraction pathways
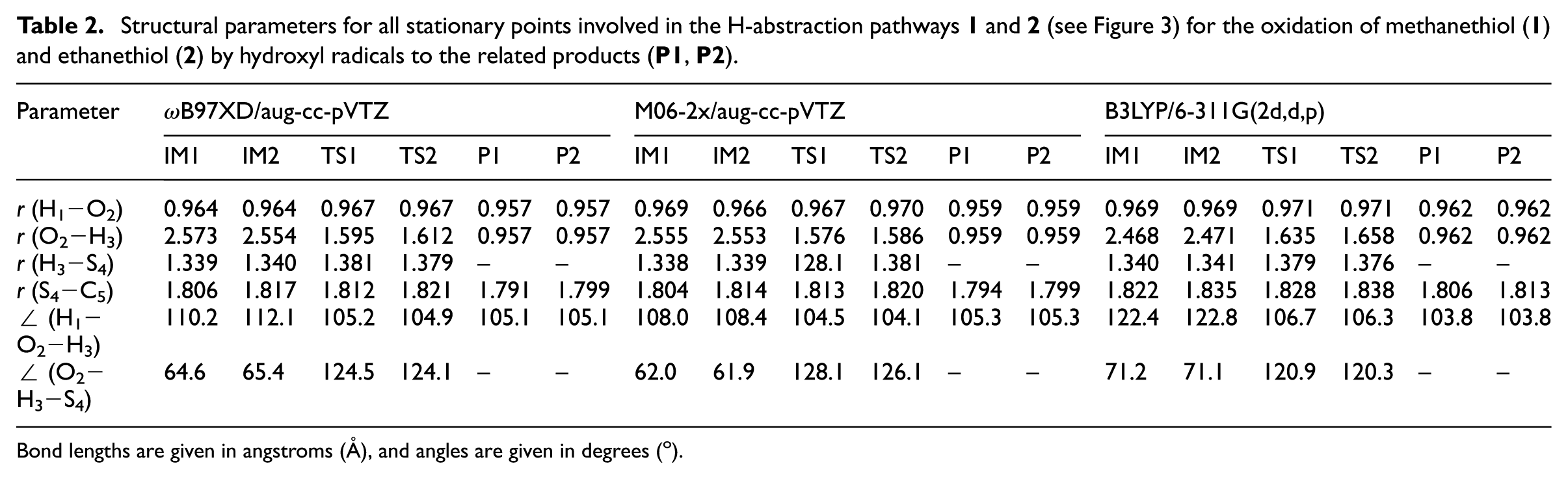
Bond lengths are given in angstroms (Å), and angles are given in degrees (o).
Structural parameters for all stationary points involved in the H-abstraction pathways
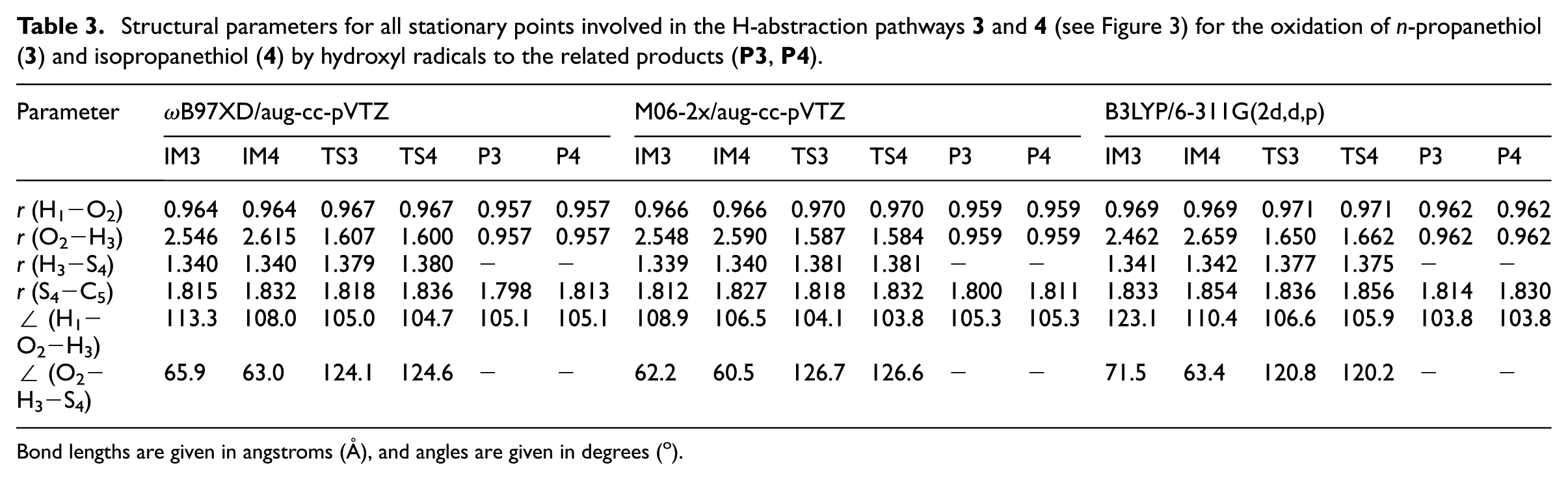
Bond lengths are given in angstroms (Å), and angles are given in degrees (o).

Potential energy profiles for the reaction pathways
According to our best CBS-QB3 data, this prereactive intermediate complex (IM
i
) for pathways
The fact that the relative elongation of the S−H bond is smaller than that inferred for the forming O−H bond indicates that the transition states (TS i ; i = 1−4) are structurally closer to the reactants than to the products. This is in line with Hammond’s principle86−89 and the observation that the removal of a hydrogen atom from the studied aliphatic thiol by a hydroxyl radical is an exothermic process.
Prior to ending this discussion of geometric structures and parameters, it is worth noting that, at all considered DFT levels of theory that were used to optimize the geometries of prereactive intermediates, TSs and products, the spin contamination [<S2>obs−0.75] never exceeds 0.011 (see Table S3 of the Supplemental Material) and can thus, for all practical purposes, be regarded as negligible. For the HF/6-31+G* wavefunction, which is required for post-self-consistent field energy calculations at the CCSD(T)/6-31+G* theoretical level in the CBS-QB3 procedure, <S2> may reach values around 0.77 for all of the identified TSs.
Energetic and thermodynamic parameters
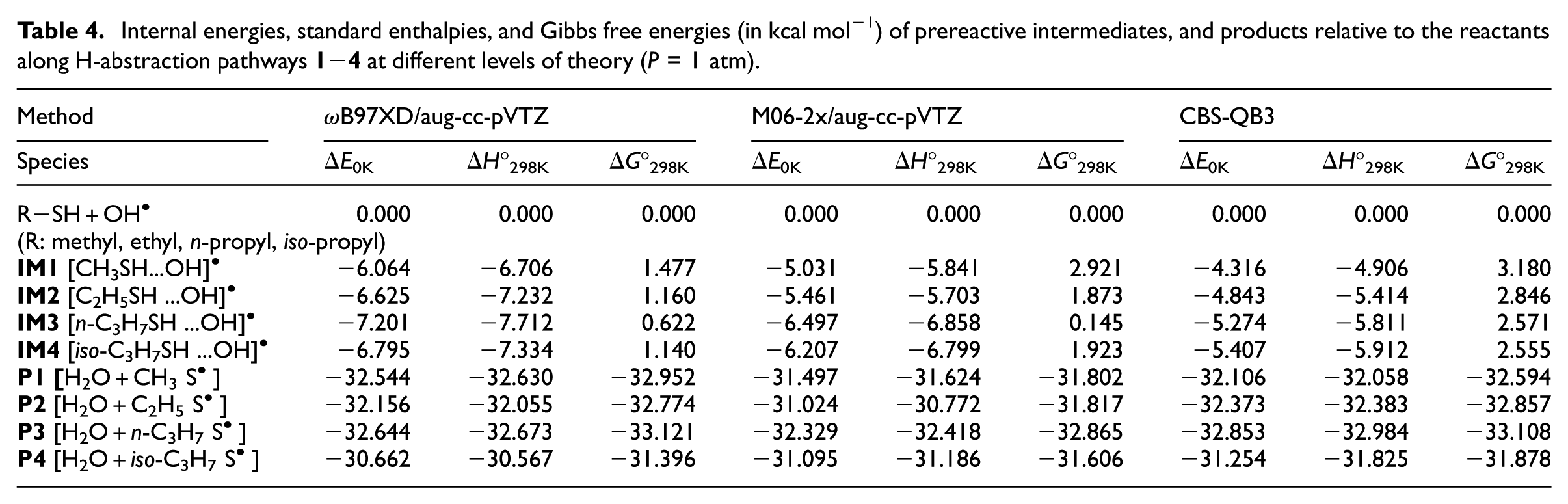
The total internal energies at 0 K, as well as the enthalpies and Gibbs free energies at 298 K of all identified intermediates, TSs, and products along pathways
Internal energies, standard enthalpies, and Gibbs free energies (in kcal mol−1) of prereactive intermediates, and products relative to the reactants along H-abstraction pathways
Internal energies, standard enthalpies, and Gibbs free energies (in kcal mol−1) of transition states relative to the reactants along H-abstraction pathways
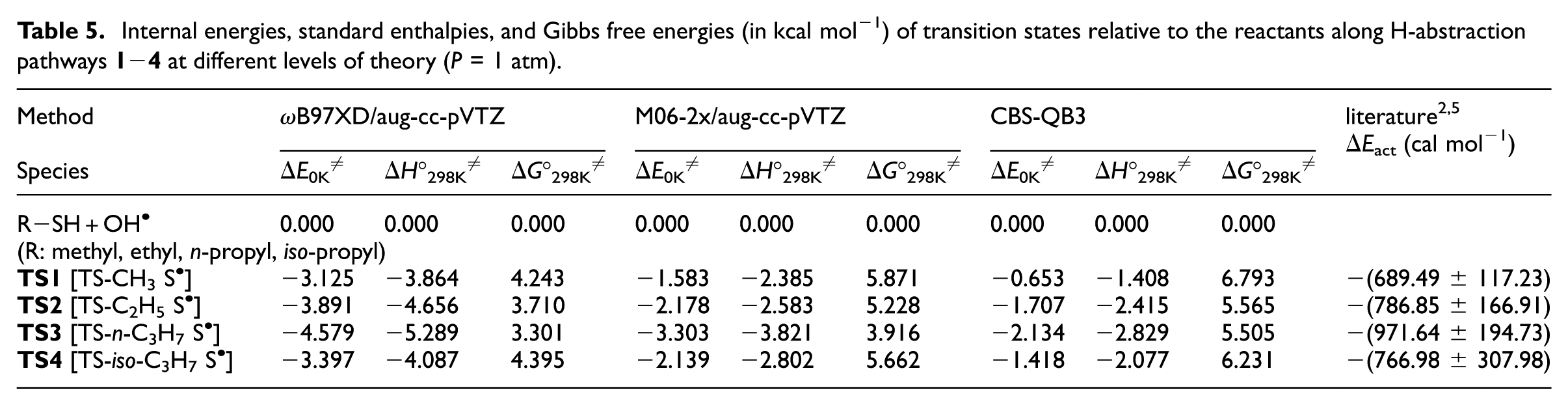
Our best (CBS-QB3) estimated value for the corresponding activation energy of these reactions is 0.65–2.13 kcal mol−1 below the reactants. According to experiment,2,5 this difference in activation energies for the bimolecular reactions R
i
+OH•→P
i
(i = 1−4) indicates that the formation of products
Reaction energies, enthalpies, entropies, and Gibbs free reaction energies for the oxidation of compounds
Comparison of the CBS-QB3 results with data obtained using ωB97XD and M06-2x exchange–correlation functionals (Tables 4 and 5) shows that the standard and widely used ωB97XD functional performs rather poorly in calculating reaction and activation energies. A much better agreement with the benchmark CBS-QB3 results is obtained with the M06-2x functional. Nevertheless, differences in activation energies of the order of 1.35 kcal mol−1 or more are still noted when comparing the ωB97XD and M06-2x results with the CBS-QB3 estimates. This, along with the extreme variability of the DFT results with respect to the employed exchange–correlation functional, shows that DFT still remains unsuited for highly quantitative studies of kinetic rate constants. 40
As can be seen in Tables 3 and 4, the newly supplied CBS-QB3 data differ sensitively from those reported by Wine et al.
2
and Atkinson et al.
5
Among all supplied data, energy values obtained with the M06-2x functional appear on average to be in closest agreement with the benchmark CBS-QB3 results. From the data provided in Table 4, we characterize in detail (Table 5) the energy barriers associated with the conversion of the prereactive intermediates IM
i
into the related products (see also Figure 3). The CBS-QB3 results show that the energy barriers (IM
i
→TS
i
; i = 1−4) encountered along the pathways
According to Hammond’s postulate, the structure of the TSs would resemble the products more than the reactants. 86 This type of comparison is especially useful because most TSs cannot be characterized experimentally. 87 Hammond’s postulate explains this observation by describing how varying the enthalpy of a reaction would also change the structure of the TS. In turn, this change in geometric structure would alter the energy of the TS and therefore also the activation energy and reaction rate. 88 This can be quantified in terms of a parameter L, defined as the ratio of the elongation of the S4−H3 bond length to the shrinkage of the H3−O2 bond distance 89

L > 1 indicates that the TS lies closer to the products, whereas L < 1 indicates that the TS lies closer to the reactants.
90
The values obtained for the parameter L for pathways
Prior to ending this discussion of reaction energies and energy barriers, it is worth noting that, at all employed levels of DFT, the spin contamination [<S2>obs− 0.75] never exceeds 0.011 (see Table S3 of the Supplemental Material) and can thus, for all practical purposes, be regarded as insignificant. As is readily apparent from Table S3 of the Supplemental Material, spin contamination of the unrestricted Hartree-Fock electronic wave functions used at the start of all CBS-QB3 computations never exceeds 0.02 in this study and can thus, for all practical purposes, be regarded as negligible. The obtained CBS-QB3 results may therefore safely be regarded as benchmark results.
Kinetic parameters
TST and RRKM estimates for individual rate constants of the studied compounds calculated in the gas phase at a pressure of 1 bar and at the considered temperatures in line with the original experiments2,5 are listed in Tables 6−9 and compared with experimental data. Further RRKM data computed at lower and higher pressures are provided for the same temperatures in Table S4a−S4i of the Supplemental Material. Theoretical values obtained using TST at a pressure of 1 bar do not differ by more than two orders of magnitude from the experimental data.
Rate constants (units: unimolecular reactions in s−1; bimolecular reactions in cm3 molecule−1 s−1) for the H-abstraction of methanethiol (compound
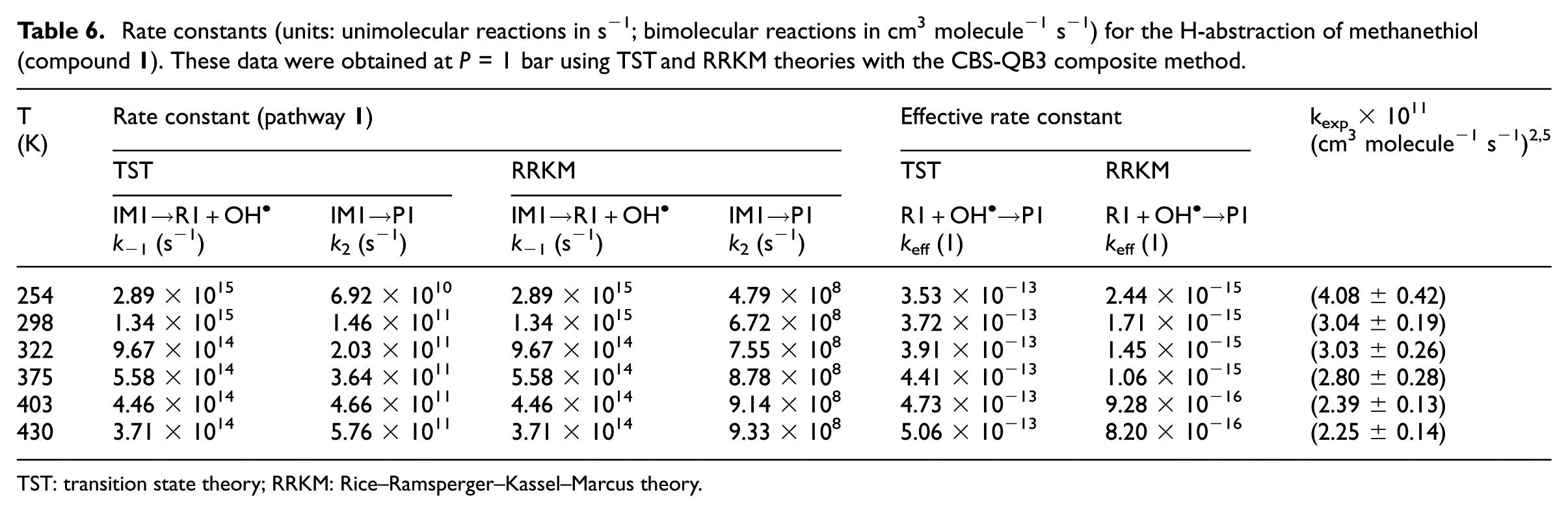
TST: transition state theory; RRKM: Rice–Ramsperger–Kassel–Marcus theory.
Rate constants (units: unimolecular reactions in s−1; bimolecular reactions in cm3 molecule−1 s−1) for the H-abstraction of ethanethiol (compound
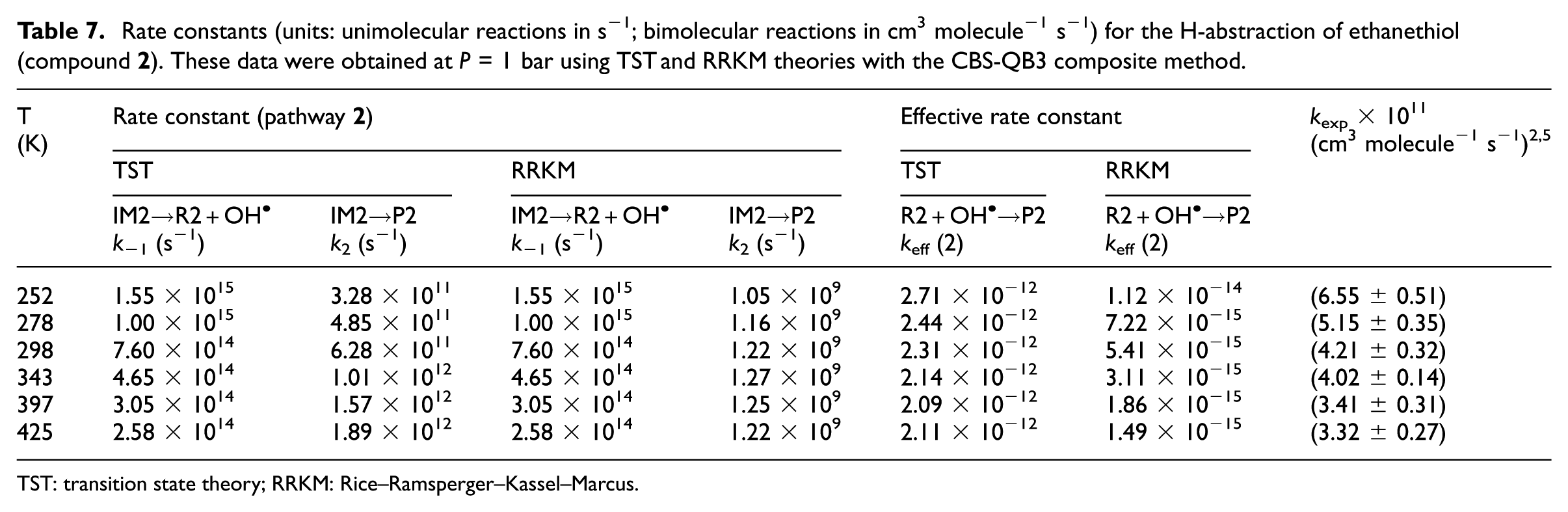
TST: transition state theory; RRKM: Rice–Ramsperger–Kassel–Marcus.
Rate constants (units: unimolecular reactions in s−1; bimolecular reactions in cm3 molecule−1 s−1) for the H-abstraction of n-propanethiol (compound
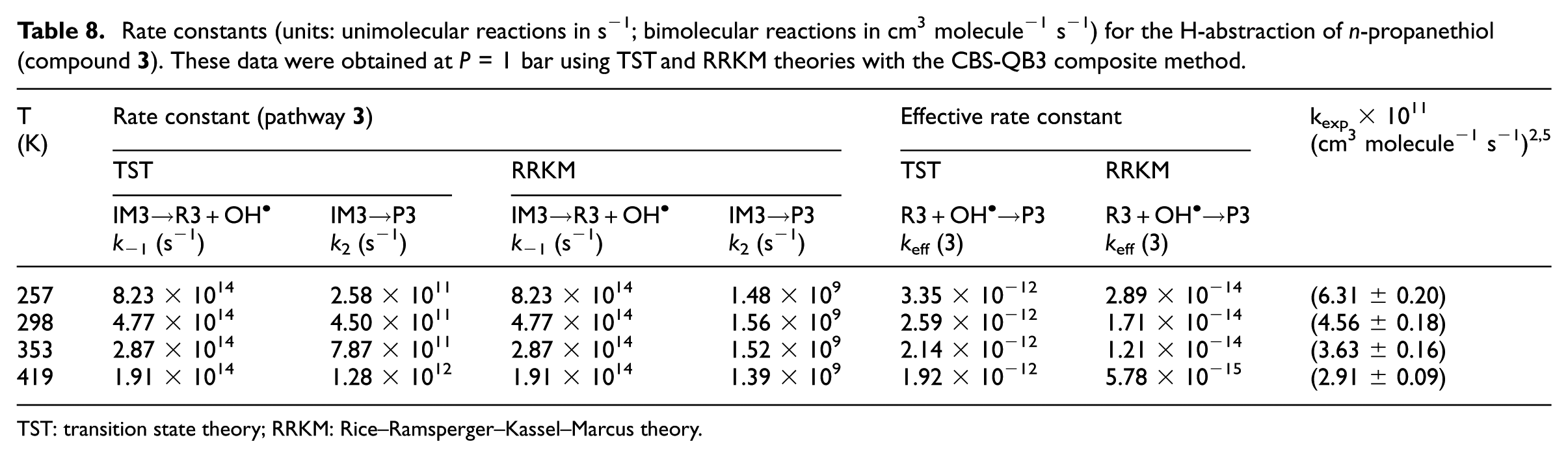
TST: transition state theory; RRKM: Rice–Ramsperger–Kassel–Marcus theory.
Rate constants (units: unimolecular reactions in s−1; bimolecular reactions in cm3 molecule−1 s−1) for the H-abstraction of isopropanethiol (compound
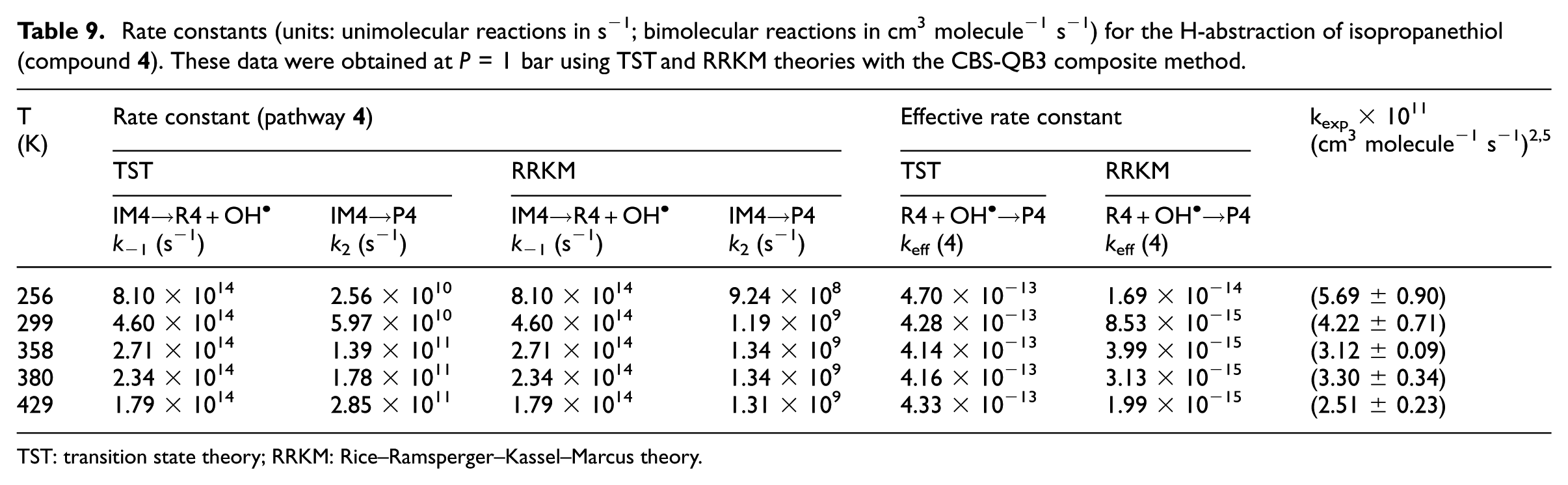
TST: transition state theory; RRKM: Rice–Ramsperger–Kassel–Marcus theory.
Effective rate constants for the reaction pathways
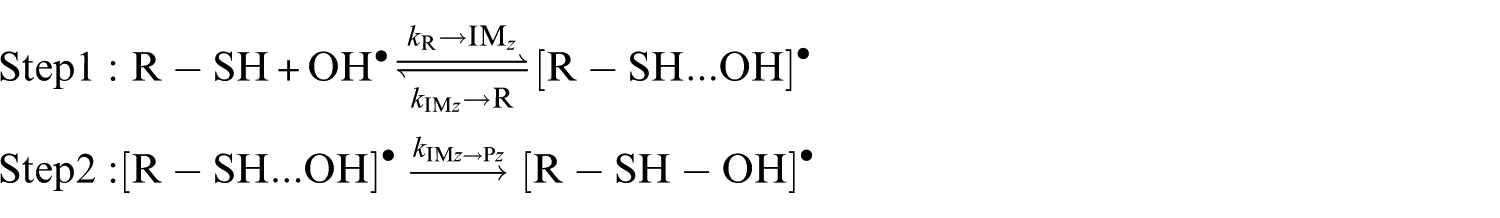
A steady-state analysis of the above sequence of reactions leads to the following easily tractable expressions for the effective rate constants characterizing the studied reaction pathways:
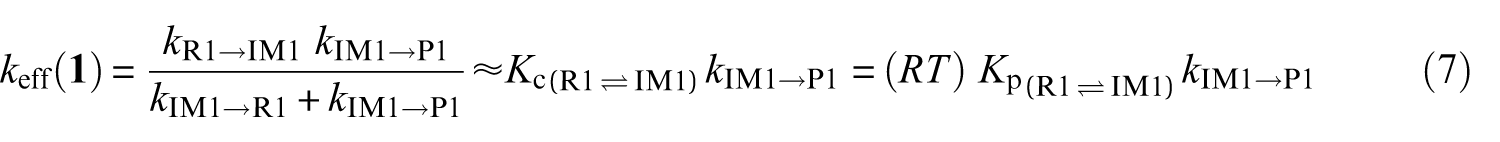
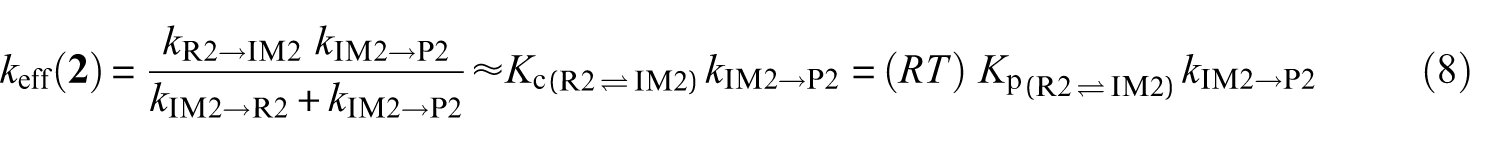
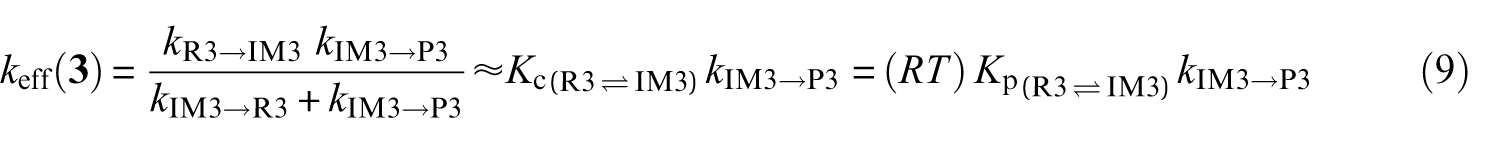
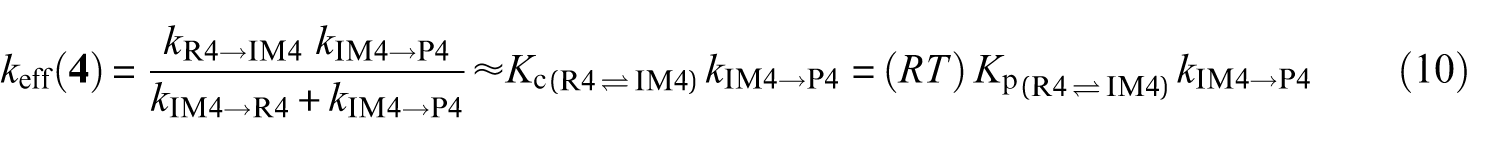
k (R→IMz,(z = 1−4)) represent the rate constants characterizing the forward bimolecular reaction steps (in cm3 molecule−1 s−1), whereas kIMz→P and kIMz→R are the forward and backward unimolecular reaction rate constants (in s−1), respectively.
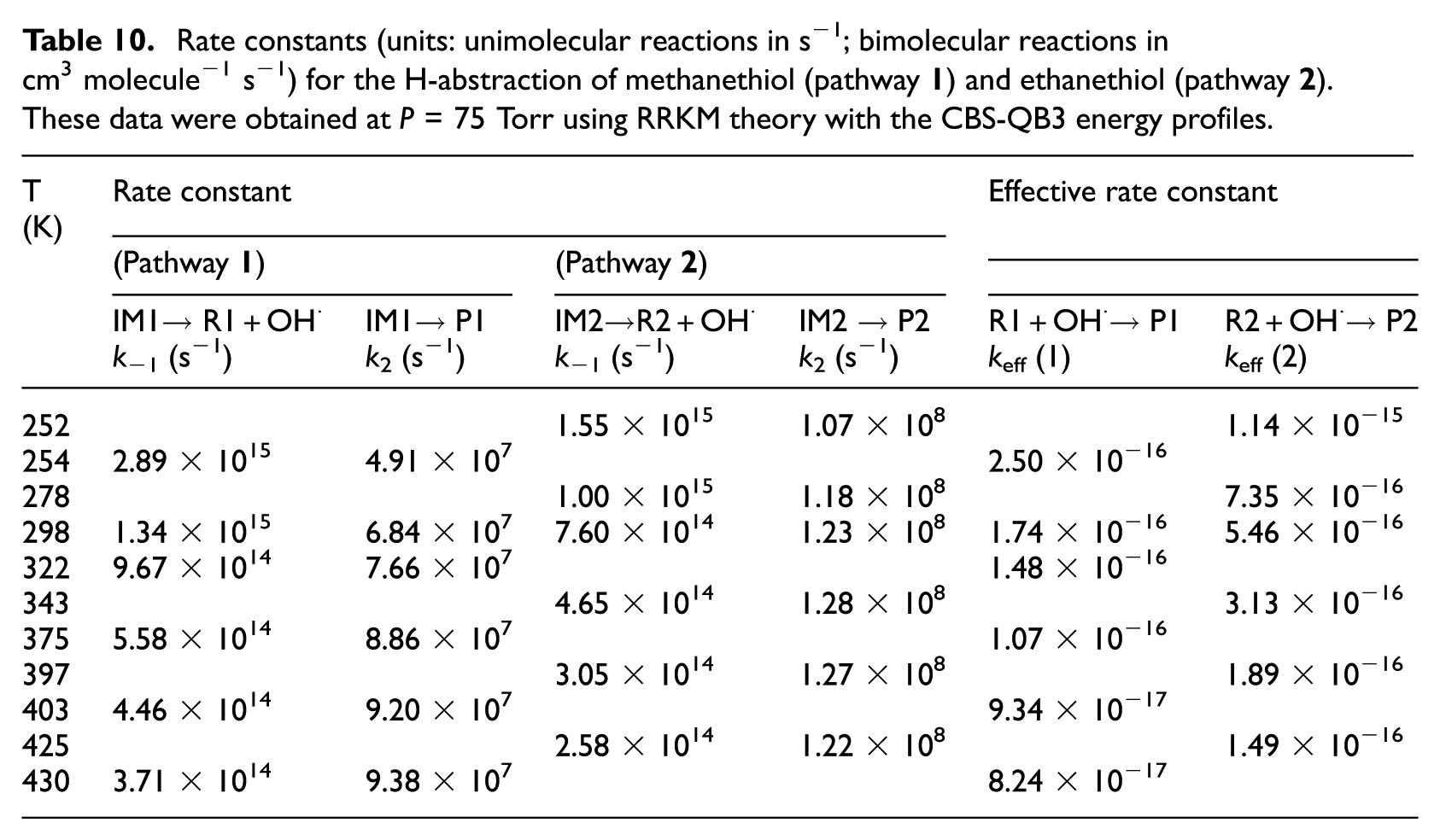
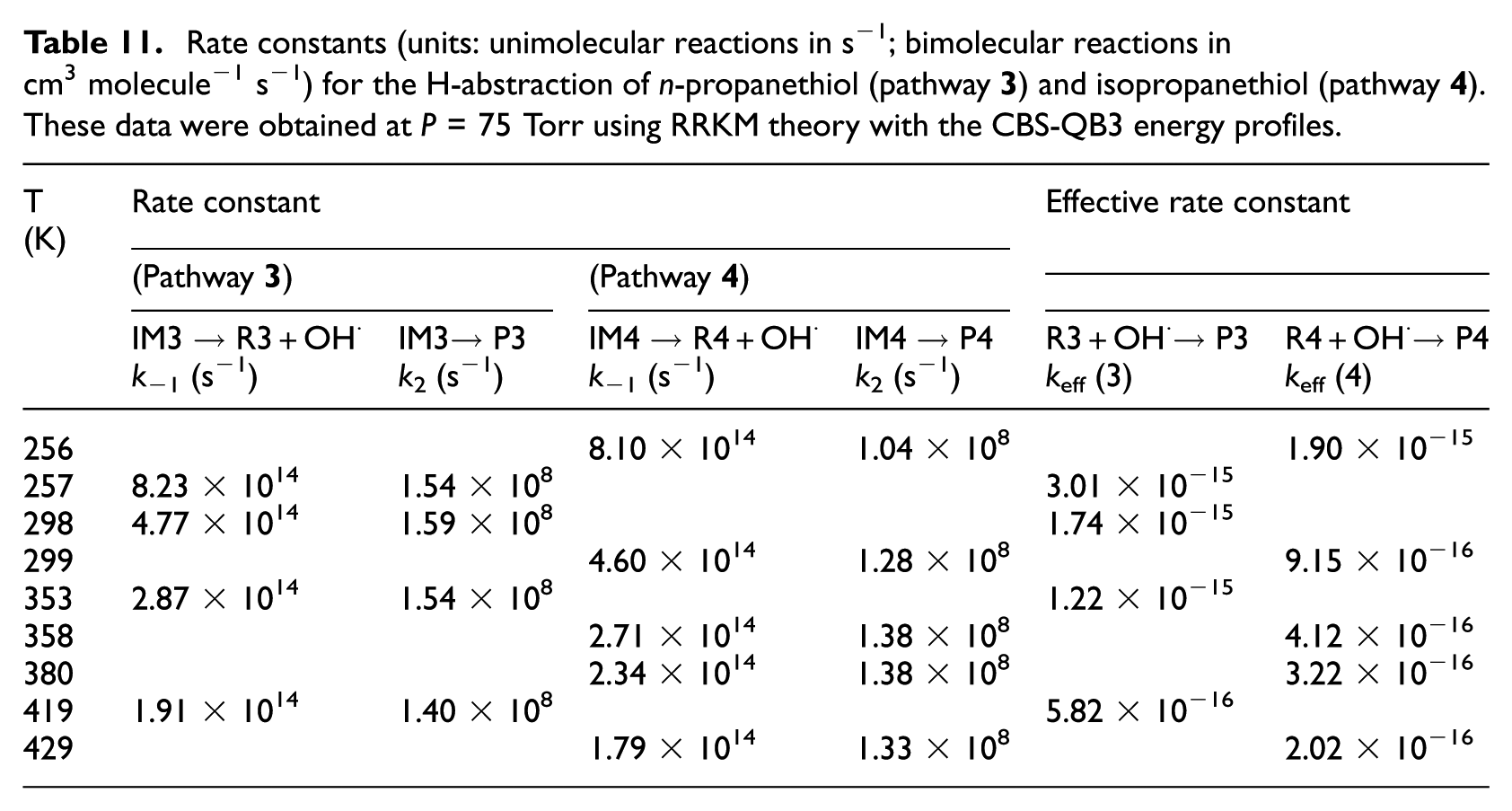
RRKM estimates for unimolecular (k2) and effective bimolecular rate constants (keff) are listed in Tables 6−9 (at P = 1 bar) and Tables 10 and 11 (at P = 75 Torr), respectively. These data were obtained from the computed CBS-QB3 energy profiles and B3LYP/6-311G(2d,d,p) microcanonical rovibrational densities of states. In Tables 6−9, theoretical rate constants can also be compared with available experimental data.2,5 These rate constants are the results of TST and RRKM calculations performed on the CBS-QB3 energy barriers and densities of states. Further RRKM data obtained at lower and higher pressures are given at the same temperatures in Table S4a−S4i in the Supplemental Material. The reader is referred further to Table S4a−S4i of the Supplemental Material for a detailed evaluation of the temperature dependence of the equilibrium constants (Kp) characterizing the first reversible reaction step.
Rate constants (units: unimolecular reactions in s−1; bimolecular reactions in cm3 molecule−1 s−1) for the H-abstraction of methanethiol (pathway
Rate constants (units: unimolecular reactions in s−1; bimolecular reactions in cm3 molecule−1 s−1) for the H-abstraction of n-propanethiol (pathway
In line with the computed energy profiles and kinetic rate constants, the formation of
Arrhenius plots (Figure 4) of the effective rate constants, which were obtained using RRKM theory for reactions
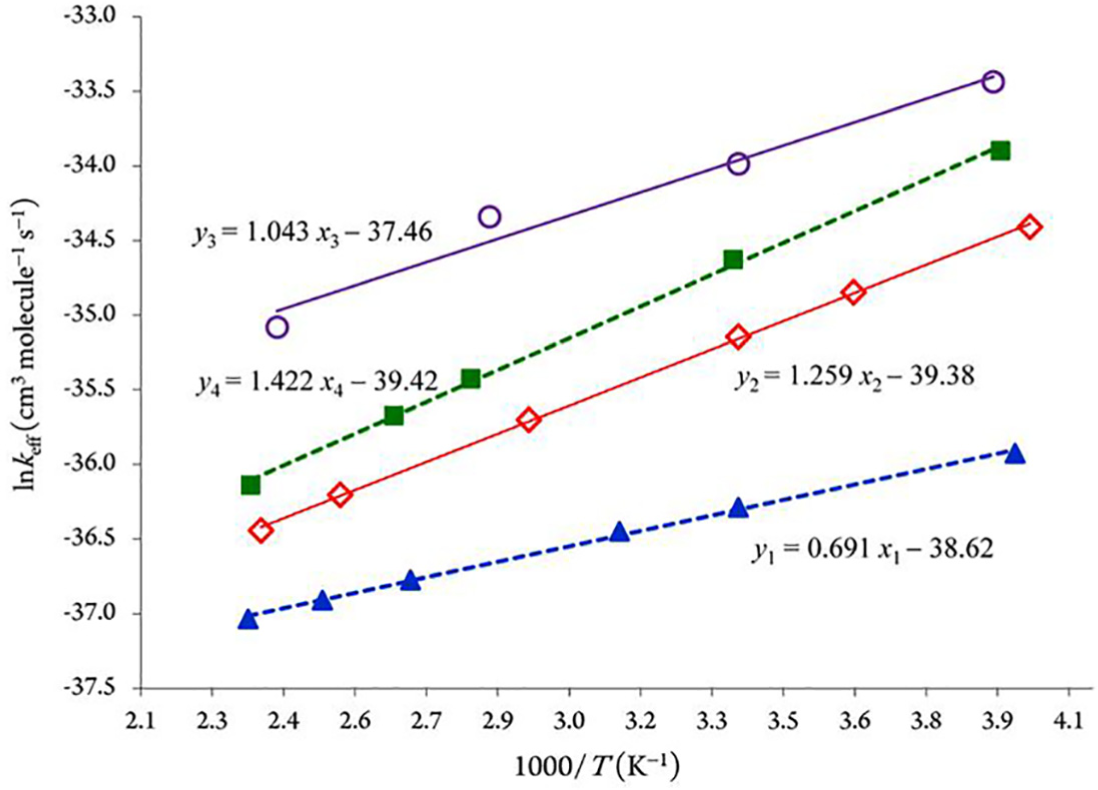
Arrhenius plots of the obtained RRKM bimolecular rate constants using the CBS-QB3 approach (P = 75 Torr).
As is to be expected, because of the negative energy barriers involved at the experimental pressure of 75 Torr,2,5 RRKM effective rate constants obtained from the CBS-QB3 energy for the fastest reaction pathway (R3+OH•→ P3) overestimate the experiments, which at the investigated temperatures (252−430 K) correspond to errors of the order of 0.04–1.16 kcal mol−1 on the computed activation energies. In view of the known performances of the CBS-QB3 approach, this semiquantitative agreement between theory and experiment is quite satisfactory, considering that hindered rotations of the OH group and anharmonic effects in the intermediates and TSs have been neglected. Furthermore, it appears quite clearly that pressures higher than ∼104 bar are required for ensuring a saturation of the computed bimolecular rate constants compared with the high-pressure limit (TST) of the RRKM bimolecular rate constants (see Figure 5). Consequently, the comparison with the RRKM data nevertheless indicates that the TST approximation breaks down at pressures higher than 104 bar for the kinetic rate constant characterizing pathway
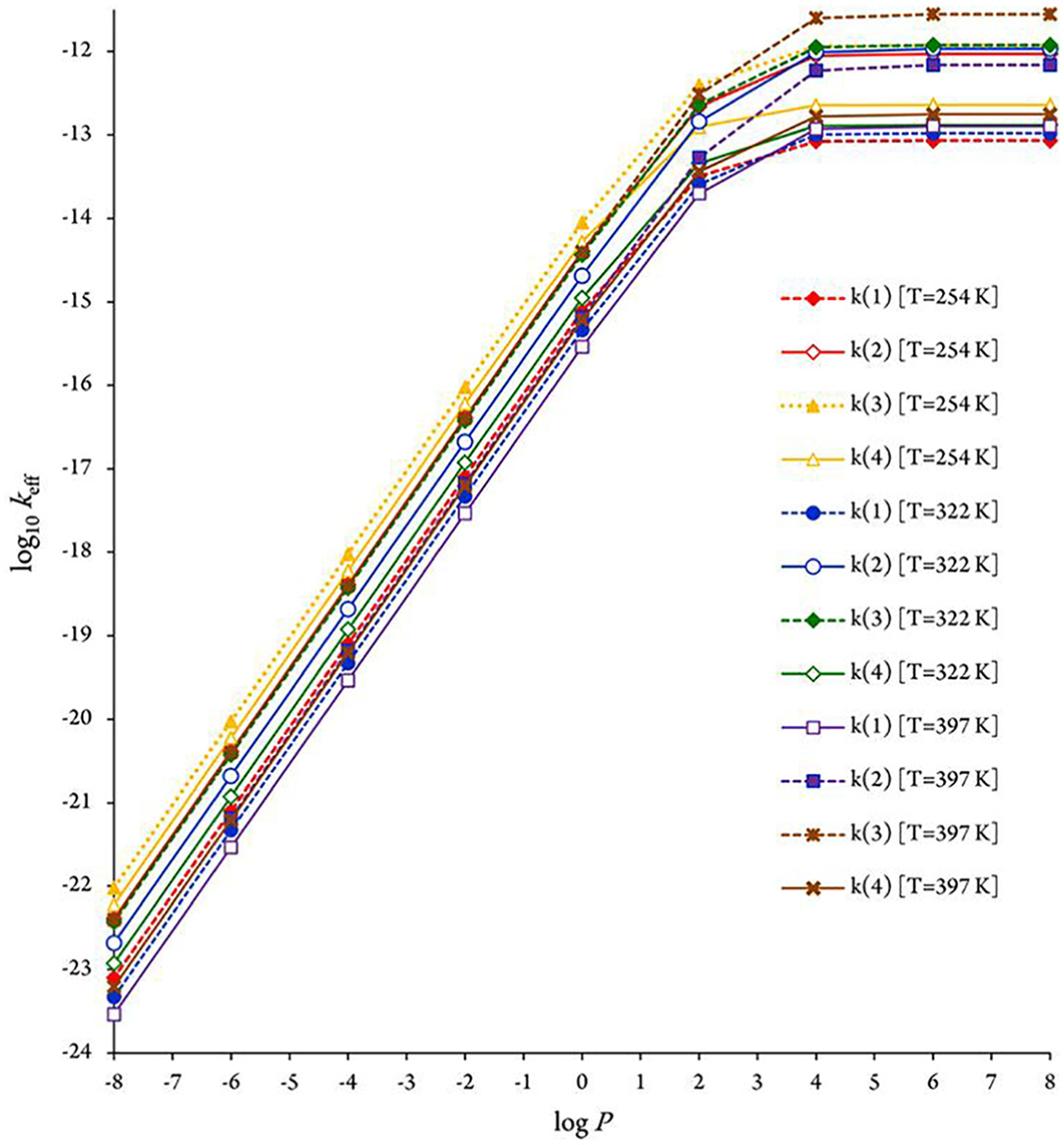
Pressure dependence of kinetic rate constants for R i →P i (i = 1−4) reaction pathways at different temperatures; results obtained with CBS-QB3 quantum chemical data.
The pressure dependence of the computed RRKM rate constants is depicted at 254, 322, and 397 K in Figure 5. Upon inspecting this figure, it is immediately apparent that rather high pressures (> ∼104 bar) are enough to ensure a saturation of the RRKM rate constants, in comparison with the high-pressure limit. This observation is most obviously the consequence of negative activation energies, which makes standard TST invalid at atmospheric pressure (1 bar). Pressure effects need therefore to be taken into account on RRKM grounds for consistent insights (Tables 10 and 11) into the experimental rate constants,2,5 which were obtained at a pressure of 75 Torr. Compared with the high-pressure limit, the pressure effects result, indeed, in this case in a most significant decrease of the computed rate constants, by approximately four orders of magnitude. These differences are due to the applied tunneling corrections.
Conclusion
The oxidation mechanisms of alkyl-substituted aliphatic thiols initiated by hydroxyl radicals in the gas phase have been studied for the first time on computational grounds using DFT along with various exchange–correlation functionals (ωB97XD and M06-2x) and the aug-cc-pVTZ basis set. The obtained reaction energies and activation barriers incorporate zero-point vibrational energy differences and counterpoise corrections for basis set superposition errors. Comparison has been made with benchmark computational results obtained at the composite CBS-QB3 level of theory. The best agreement with the computed energy barriers is obtained with the M06-2x exchange–correlation functional. Kinetic rate constants for unimolecular and bimolecular reaction steps were estimated by means of TST and statistical RRKM theory.
These first reaction steps for OH addition in pathways
Effective rate constants have been calculated according to a steady-state analysis of a two-step model reaction mechanism, assuming reversibility of the first bimolecular addition reaction step, and irreversibility of the second unimolecular dissociation step. In line with the experimental observations,2,5 results indicate that the kinetically most efficient process at temperatures ranging from 252 to 430 K corresponds to OH addition onto n-propanethiol (
In line with negative activation energies, 91 it was found that the standard TST approximation breaks down at ambient pressure (1 bar) for the first bimolecular reaction steps. RRKM calculations show in particular that overwhelmingly high pressures, higher than 104 bar, are required for restoring of this approximation for all reaction channels involved in the oxidation of the studied pathways, in particular for the conversion of the prereactive van der Waals complex [R−SH…OH]• into the molecular energized adduct [R−SH-OH]•.
A comparison with TST results seems to validate RRKM theory for all studied reaction pathways. The RRKM theory appears to be sufficient for achieving semiquantitative insights into the experimentally kinetic rate constants, with discrepancies in the range of three to four orders of magnitude between theory and experiment.
Note that the oxidation reaction mechanisms of the alkyl-substituted aliphatic thiols involved in the so-called OH addition pathways (the OH radical attacks the sulfur atom) were the subject of the separate theoretical study using a similar DFT and CBS-QB3 quantum chemical approach that has been published. 92
Supplemental Material
Electronic_Supplementary_Information – Supplemental material for Oxidation reaction mechanism and kinetics between OH radicals and alkyl-substituted aliphatic thiols: H-abstraction pathways
Supplemental material, Electronic_Supplementary_Information for Oxidation reaction mechanism and kinetics between OH radicals and alkyl-substituted aliphatic thiols: H-abstraction pathways by Arezoo Tahan and Abolfazl Shiroudi in Progress in Reaction Kinetics and Mechanism
Footnotes
Acknowledgements
This article has been derived from the research project entitled “Theoretical study on mechanisms and kinetics of the gas phase reactions of the hydroxyl radicals with aliphatic thiols containing short chain alkyl groups (1 to 3 carbon atoms).” The authors thank the anonymous referees for their highly relevant comments.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was financially supported by the Semnan Branch, Islamic Azad University, through Grant No. 17752.
Supplemental material
The Supplemental Material (Tables S1–S4) for this article can be found in the online version. Table S1: Temperature dependence of the equilibrium constants for the first bimolecular reaction step [R
i
+OH•.→IM
i
; i = 1–4] along the pathways
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.