
Abstract
Kinetic rate constants for the oxidation reactions of OH radicals with CH3SH (
Keywords
Introduction
The emission of gaseous sulphur compounds into the atmosphere occurs from both natural and anthropogenic processes. 1 Despite their low concentrations, larger organic sulphur compounds may play an important role in tropospheric sulphur chemistry. Kinetic and mechanistic data for the reactions of reduced-sulphur compounds (such as aliphatic thiols, R−SH) with hydroxyl radicals are required to evaluate their residence times and their ultimate fates in the atmosphere.2–11 Oxidation processes are thought to be the main atmospheric sink of aliphatic thiols (R−SH, R: CH3, C2H5, n-C3H7, iso-C3H7), which play a dominant role in the troposphere3,12–20 and their rate constants are, thus, needed in order to understand their involvement in photochemical air pollution. Since there are no reported theoretical data for these reactions, in this work, absolute kinetic rate constants for the reaction of OH radicals with the studied compounds have been determined over the temperature range 252−430 K.
The reaction of hydroxyl radicals with CH3SH has been studied experimentally by Wine et al.,
21
Atkinson et al.,
2
MacLeod et al.
22
and Lee and Tang
4
by the flash photolysis-resonance fluorescence technique, while Cox and Sheppard
5
used a relative rate technique. They suggested that the kinetic rate constants obtained through this oxidation reaction were pressure-dependent. Atkinson et al.
2
as well as Wine et al.
3
have reported a negative activation energy for the OH+CH3SH reaction. Wine et al.
21
concluded that addition to the sulphur atom is the dominant pathway for reactions between OH• radicals and the studied compounds, which is in agreement with the preceding study by Hatakeyama and Akimoto.
23
The obtained rate constants for compounds
Hydrogen abstraction from the carbon atom
Hydrogen abstraction from the sulphur atom
OH addition to the sulphur atom
The kinetic rate constants obtained for the studied reactions are nearly independent of the nature of the hydrocarbon chain, which clearly indicates at 298 K that hydrogen abstraction from a carbon atom is a negligible reaction channel. 21 Thus, reactions between R−SH and hydroxyl radicals must proceed through either hydrogen abstraction from the weak S−H bonds or by the formation of [R-SH−OH]• adducts.24,25
In line with our preceding article, which has been accepted for publication in this journal, 26 we have studied oxidation processes of aliphatic thiols containing short-chain alkyl groups (1−3 carbon atoms) by hydroxyl radicals through OH attack onto the hydrogen bonded to the sulphur atom to yield a water molecule and the related radicals. Based on several previous experiments which were carried out in this regard,2–4,5,21–23,27–29 we have studied in the second part of our research the involvement of an OH•radical attack on the sulphur atom, which dominates under inert (Ar) conditions, leading to the related energized adducts [R-SH−OH]• followed by hydrogen atom elimination, and the preceding experiment shows that the OH-addition reaction mechanism is believed to dominate. In this regard, Hatakeyama and Akimoto 23 suggested that CH3 S• species is produced by thermal decomposition of the [CH3SH−OH]• adducts under atmospheric conditions.
The experimental measurements2,21 of the kinetic rate constants for the reaction between OH radicals and compounds
To explain the disparity of the kinetic data supplied in Scheme 1, chemical pathways
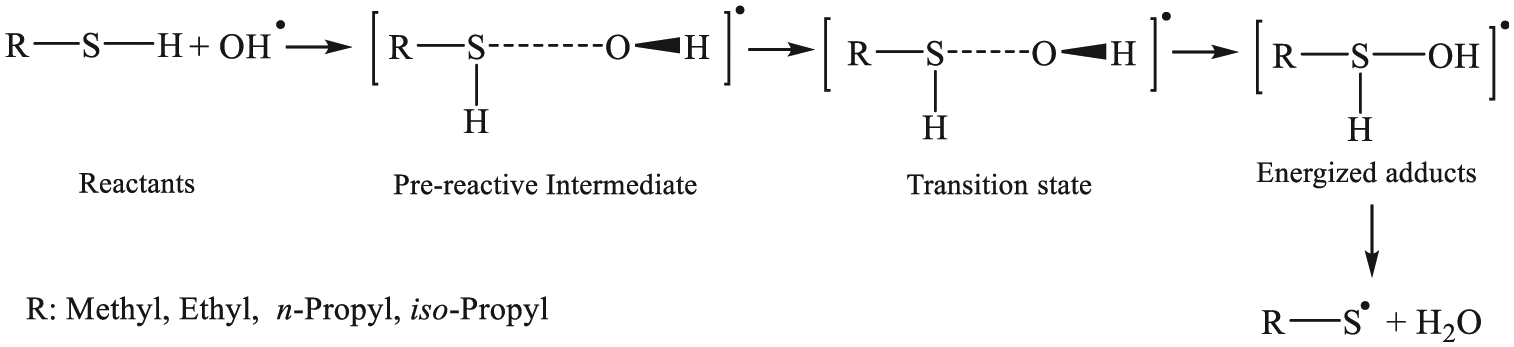
OH-addition pathways of aliphatic thiols by hydroxyl radicals.
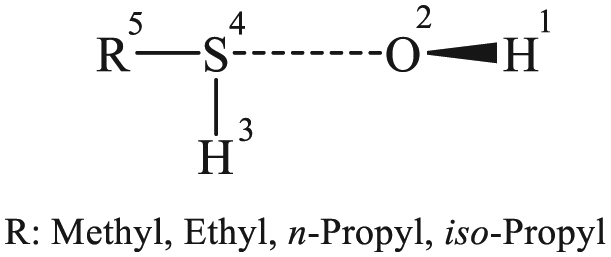
Retained atom labelling for characterizing the structures of intermediates and transition states during the oxidation of the studied thiols by OH radicals.
To gain more quantitative insights into these reaction mechanisms, we calculated reaction energies and energy barriers by the CBS-QB3 composite method.30–38 The aim of this work was to achieve comprehensive and quantitative theoretical insights of the studied pathways. Furthermore, kinetic rate constants in the high pressure limit were to be determined using transition state theory (TST),39–45 and their falloff behaviour was to be investigated at lower pressures by means of statistical Rice–Ramsperger–Kassel–Marcus (RRKM) theory46,47 to unravel the available experimental data at temperatures ranging from 252 to 430 K.
Theory and computational details
All the calculations reported in this study were performed using the Gaussian 09 package of programmes. 48 The high-level composite method CBS-QB3 has been used in this work. Analysis of vibrational frequencies confirmed that all transition structures have one imaginary frequency. Vibrational frequencies calculated at the CBS-QB3 theoretical level were used for characterization of stationary points as minima and transition states, for zero-point energy (ZPE) corrections. Intrinsic reaction coordinate (IRC) calculations 49 have been systematically performed at the B3LYP/6-311G(2d,d,p) level of theory to ensure that the computed transition structures connect the desired reactants and products.
The relative energies (the total energy of the reactants was set to zero for reference) of all species which were involved in the oxidation of compounds
In our study, reactions between aliphatic thiols and OH• radicals were found to be consistent with the following scheme 51 which assumed that this reaction occurs according to a two-step mechanism, 52 involving first a fast pre-equilibrium between the isolated reactants (R−SH+OH•) and a pre-reactive complex [R−SH….OH]• (IM i ), and thence to the related products:

where k1 and k−1 are the forward and reverse reactions associated with the first step, whereas k2 is the rate constant corresponding to the second step. The overall rate constant is given by:
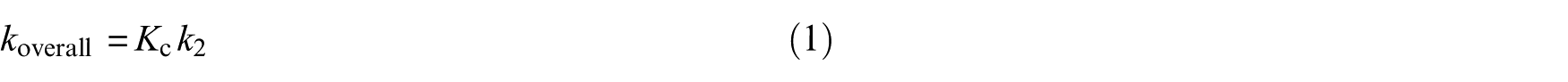
where Kc = k1/k−1 is the equilibrium constant for fast pre-equilibrium between the isolated reactants and the prereactive complex (see Table S1 of the Electronic Supplementary Information (ESI)). In the high-pressure limit (TST), kinetics characterizing the unimolecular dissociation reaction are given by39–46,53
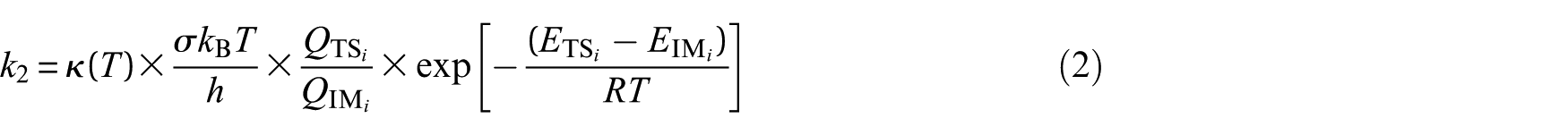
where the meaning of all involved parameters was illustrated in a prior paper. 26 Kinetic rate coefficients for the studied pathways were calculated over temperature ranges 252−430 K and in the high-pressure limit (1 atm) using TST. The overall rate constant is given by54,55
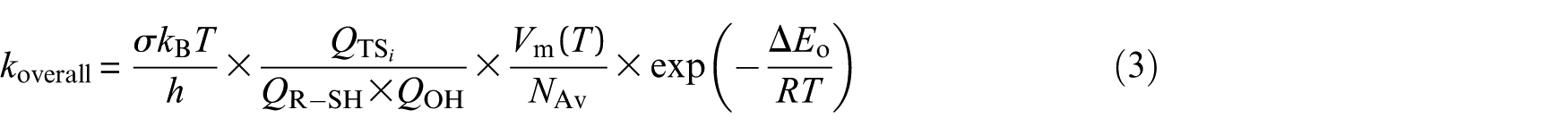
where σ is the reaction-path degeneracy, QR−SH, QOH and QTS i represent the total molecular partition functions for the isolated reactants and transition state (TS i ), respectively, and κ(T) denotes the Wigner tunnelling correction factor (see Table S2 of the ESI).56,57
Since the computed energy differences account for ZPVEs, vibrational partition functions were computed using the vibrational ground state as energy reference. TST gives an estimate of the upper-limit for rate constants as a function of temperature and is known to give reliable estimations of rate constants47,58 in the high pressure limit, especially for cases with significant barrier heights.
The prevailing pressures were measured to be enough in order to reliably calculate kinetic rate constants by means of TST. The fall-off behaviour of canonical kinetic rate constants k(T) from the TST limit (P→∞) towards the low-pressure limit (P→0) was also calculated using RRKM theory.59–61 The microcanonical kinetics k(E) were evaluated according to unimolecular RRKM theory 62

where ρ(E) is the density of states of the reactants and N†(E) denotes the total number of states at the transition state.
Kinetic rate coefficients (k) of pathways
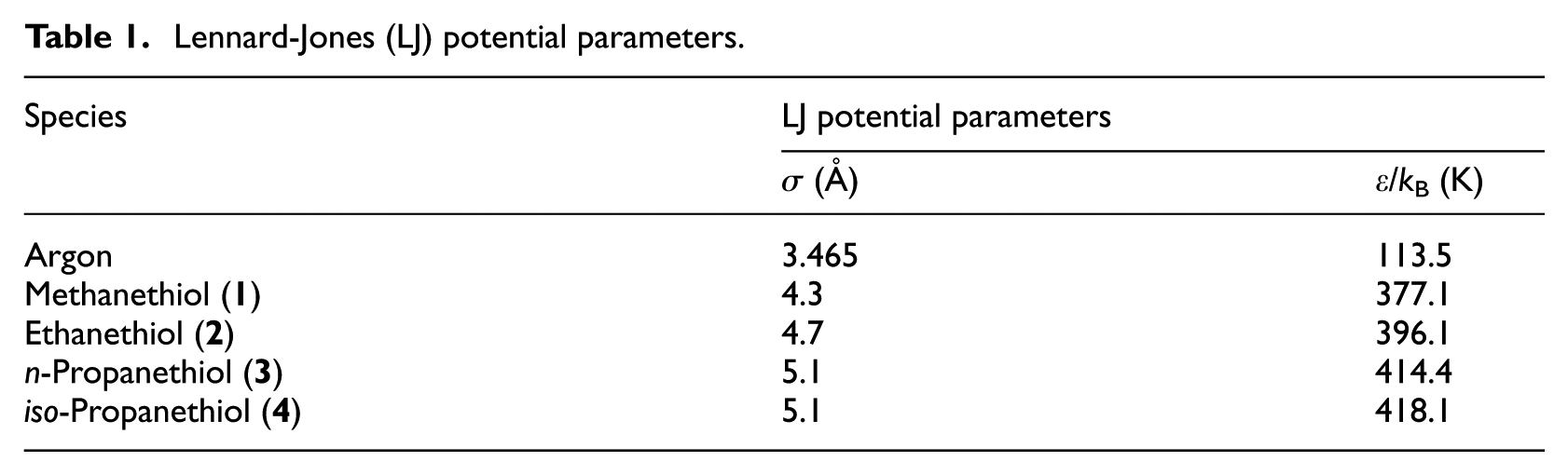
Lennard-Jones (LJ) potential parameters.
Results and discussion
Structural characteristics of stationary points
The optimized molecular structures of the intermediate complexes, transition states and products in the reactions between compounds
Structural parameters for all stationary points which are involved in the reaction between the studied aliphatic thiols and OH radicals using the CBS-QB3 approach.
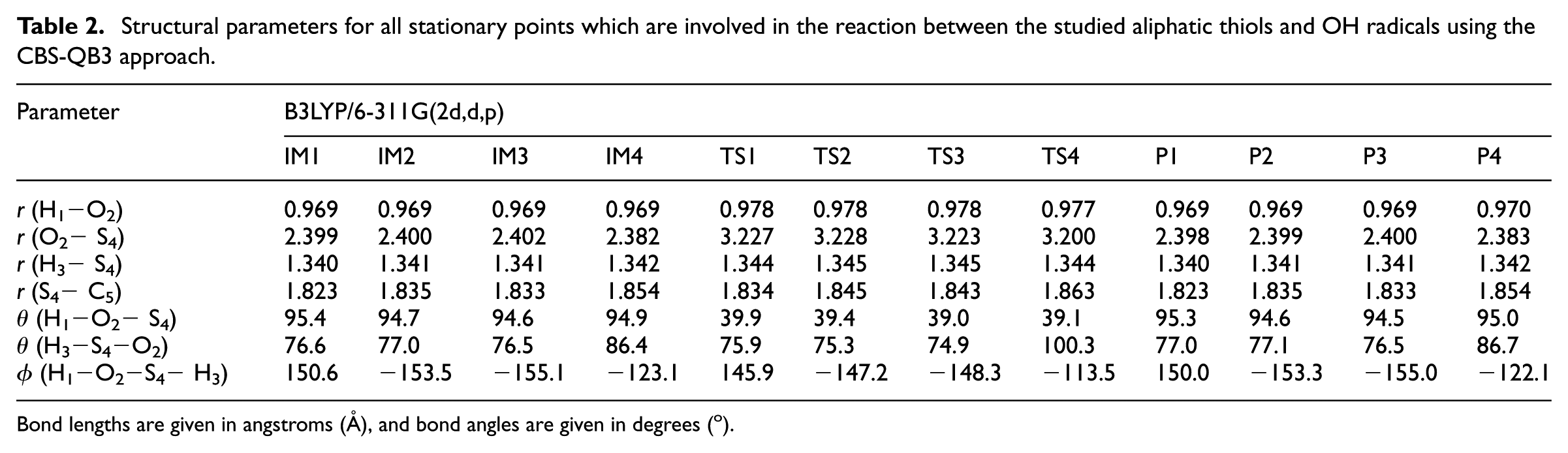
Bond lengths are given in angstroms (Å), and bond angles are given in degrees (o).
In line with changes in chemical bond orders, examination of Table 2 and comparison of the structures of the intermediates of pathways
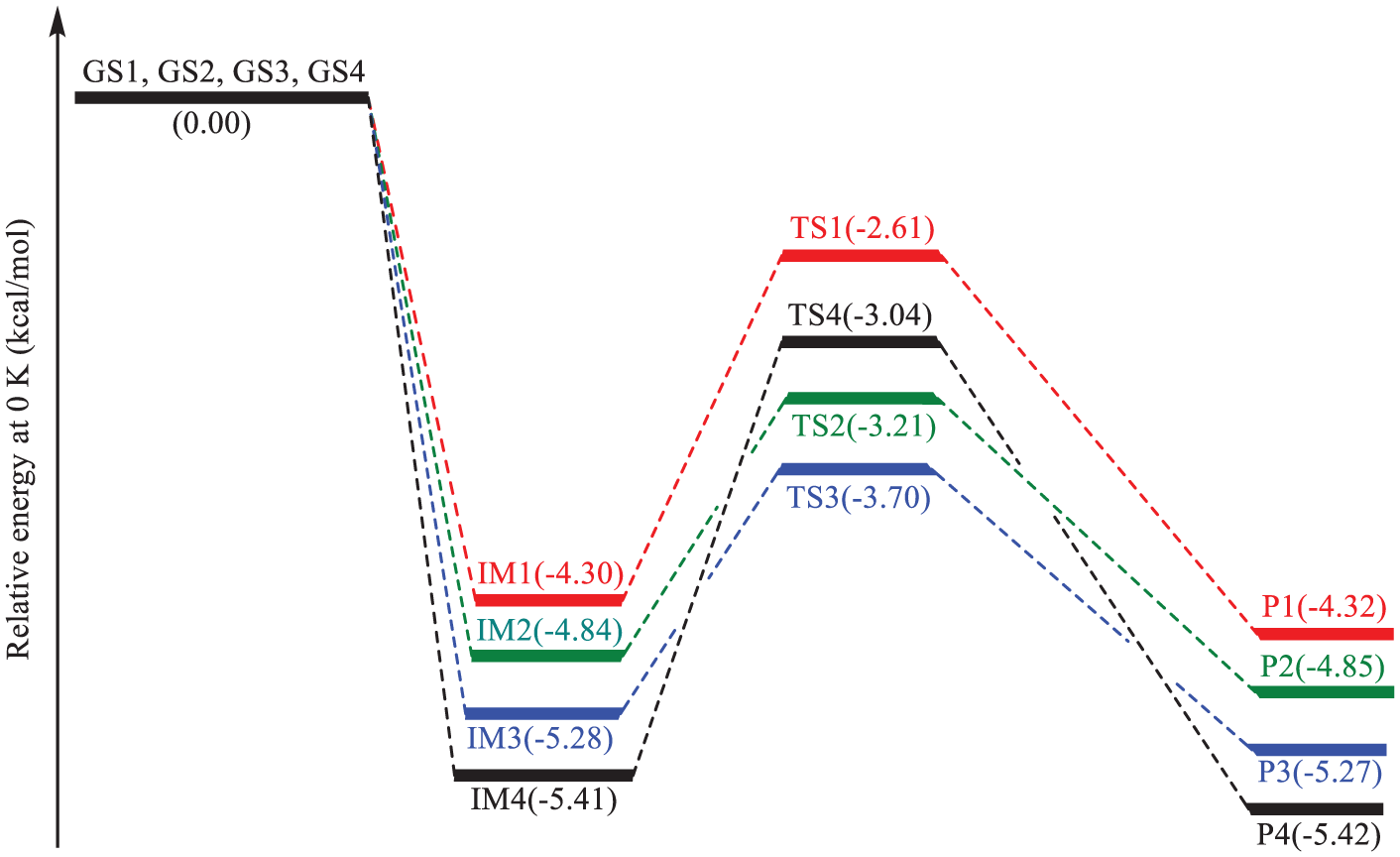
Potential energy profile for the OH-addition pathways
Most significant differences are found for the O2− S4‘bond’ length in the IMs and TSs involved in the studied pathways (Table 2). These pre-reactive intermediates are loosely bound complexes which are held mainly by van der Waals (dispersion) forces. The extreme looseness of this complex and of the associated transition state explains the disparity of the computed bond lengths. Since van der Waals forces are weak, it is very unlikely that improvements of the geometry will strongly affect the activation and reaction energies characterizing this profile.
Prior to ending this discussion of geometric structures and parameters, it is worth noticing that for the HF/6-31+G* wavefunction which is required for post-self-consistent field energy calculations at the coupled cluster theory along with single, double and perturbative triple electronic excitations/6-31+G* level of theory in the CBS-QB3 procedure, the < S2 > operator may reach values around 0.76 for all of the identified transition states (see Table S3 of the ESI).
Energetic and thermodynamic parameters
The total internal energies at 0 K, as well as the enthalpies and Gibbs free energies at 298 K, of all identified intermediates (
Energy changes along the OH-addition pathways
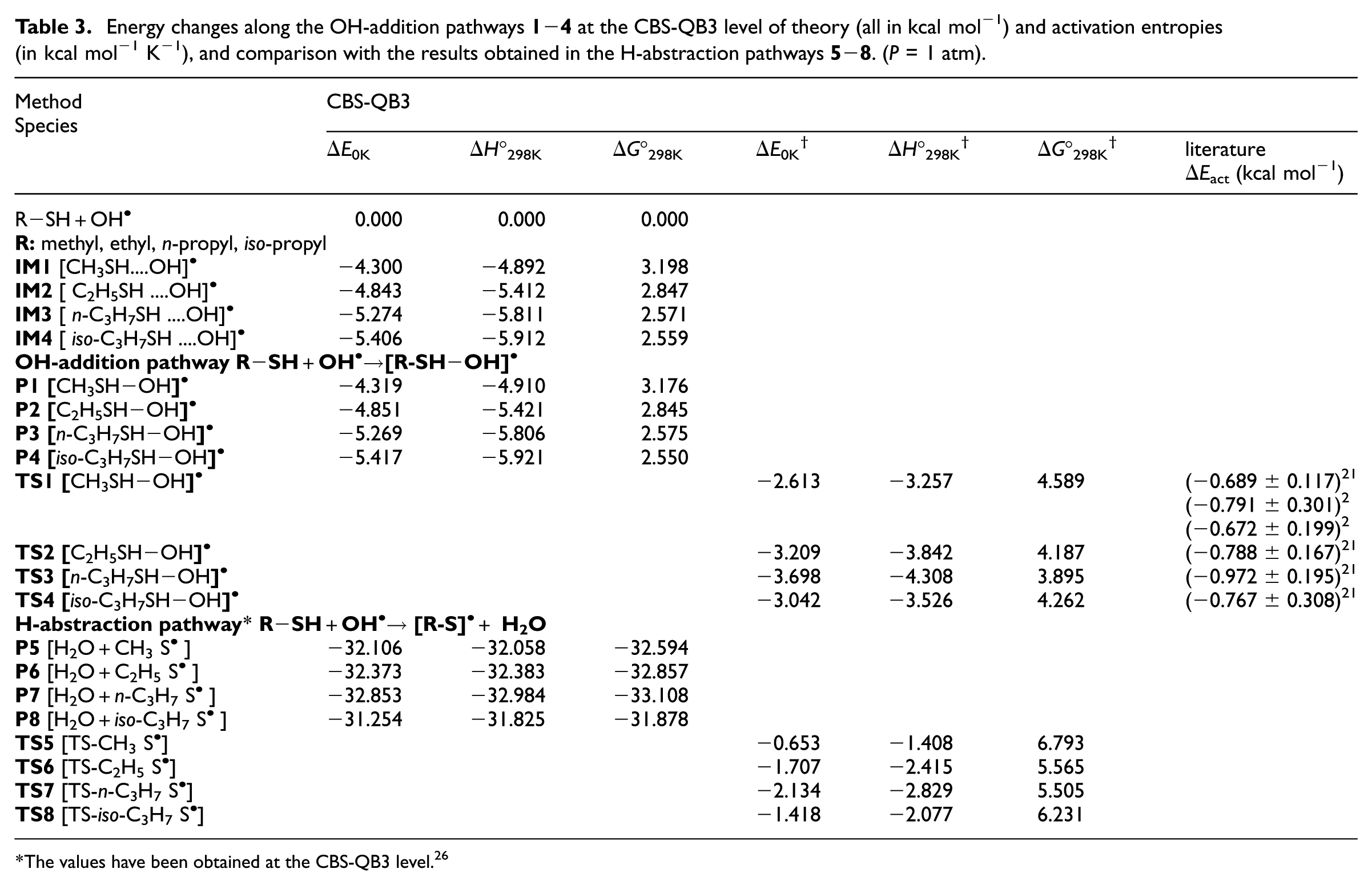
The values have been obtained at the CBS-QB3 level. 26
According to experiment,2,21 this difference in activation energies for the bimolecular reactions R
i
→P
i
(i = 1−4) indicates that the formation of product
Rate constants (units: unimolecular reactions in s−1; bimolecular reactions in cm3 molecule −1 s−1) for the reaction of OH radicals with CH3SH (compound
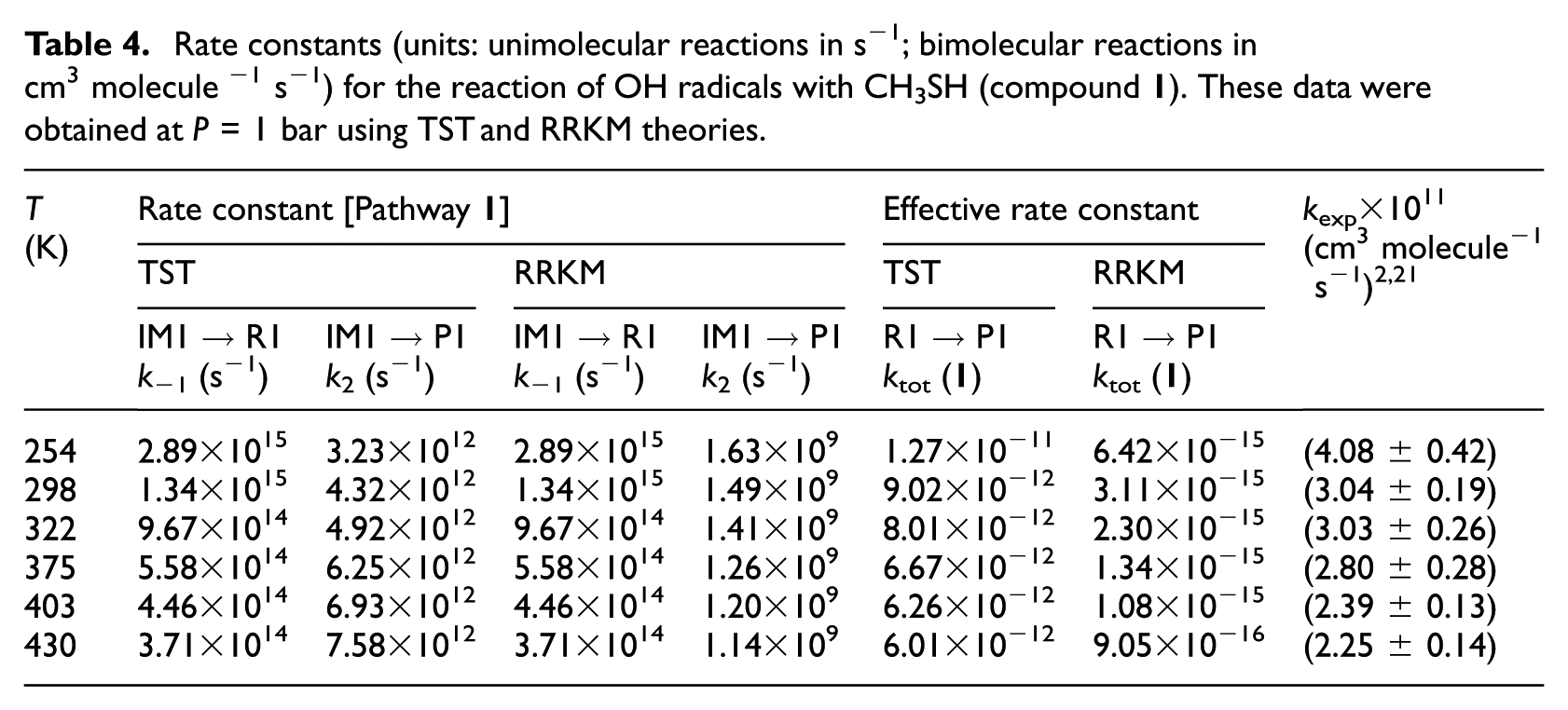
As can be seen in Table 3, the newly supplied CBS-QB3 data differ sensitively from those reported by Wine et al.
21
as well as Atkinson et al.
2
From the data summarized in Table 3, we have characterized in detail the energy barriers associated with the conversion of the pre-reactive intermediate IM
i
into the related products (
The difference in reaction energies for both oxidation reactions (OH-addition and H-abstraction pathways) indicates that formation of products (
Furthermore, as can be seen in Table 3, the difference in activation energies for both the bimolecular reactions indicates that the formation of products
Based on Hammond’s postulate, the location of the transition state structure along the reaction coordinate is given by nT parameter defined as follows:67,68

where nT represents the degree of similarity between the transition state and the product. If nT < 0.5, the TS structure is similar to that of the reactants (early TS), and if nT > 0.5, it is similar to the products (late TS). Based on the CBS-QB3 estimates, for the IMi→Pi (i = 1−4) reaction steps, nT is found to be equal to 0.496, 0.5, 0.499 and 0.498, respectively, which implies that the TS structures are located almost halfway between the pre-reactive complexes and the products.
Kinetic parameters
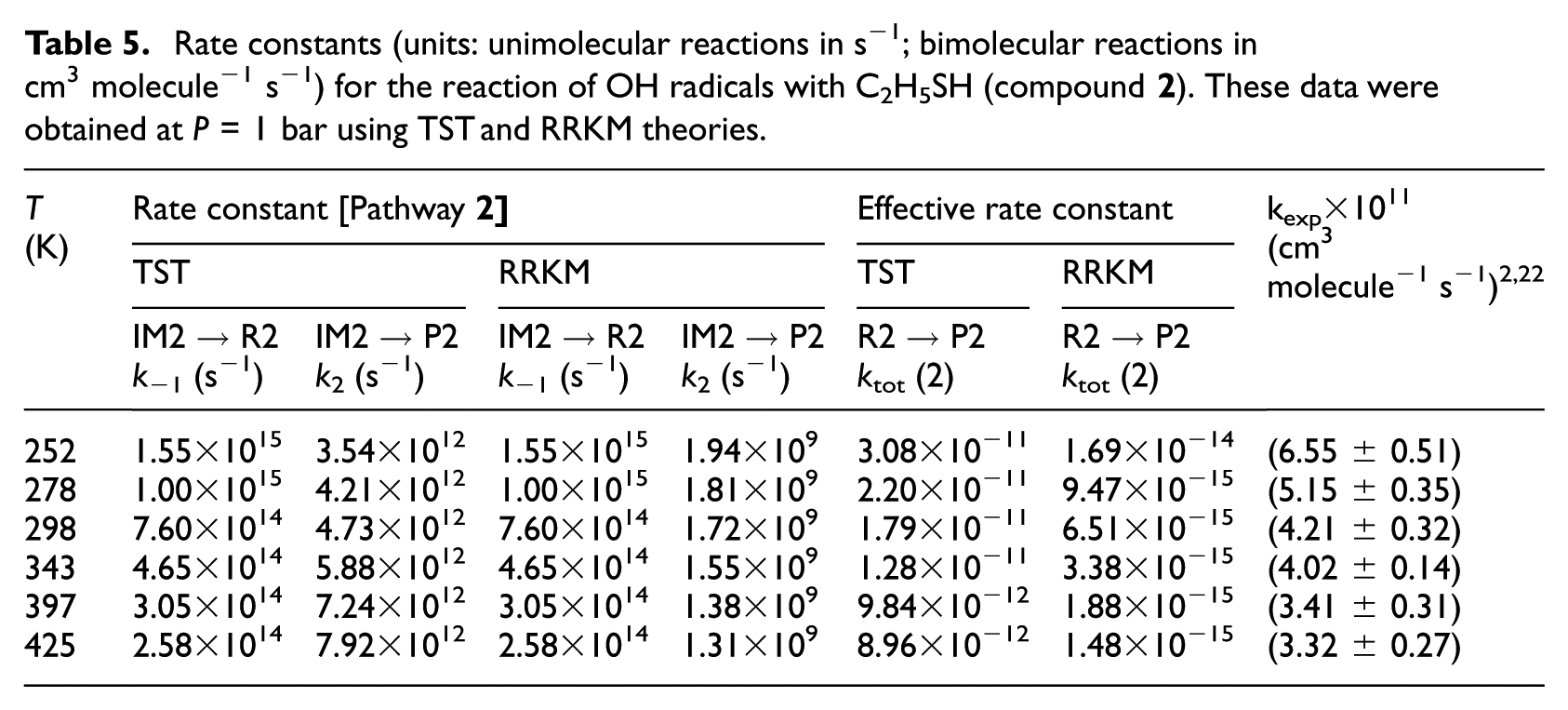
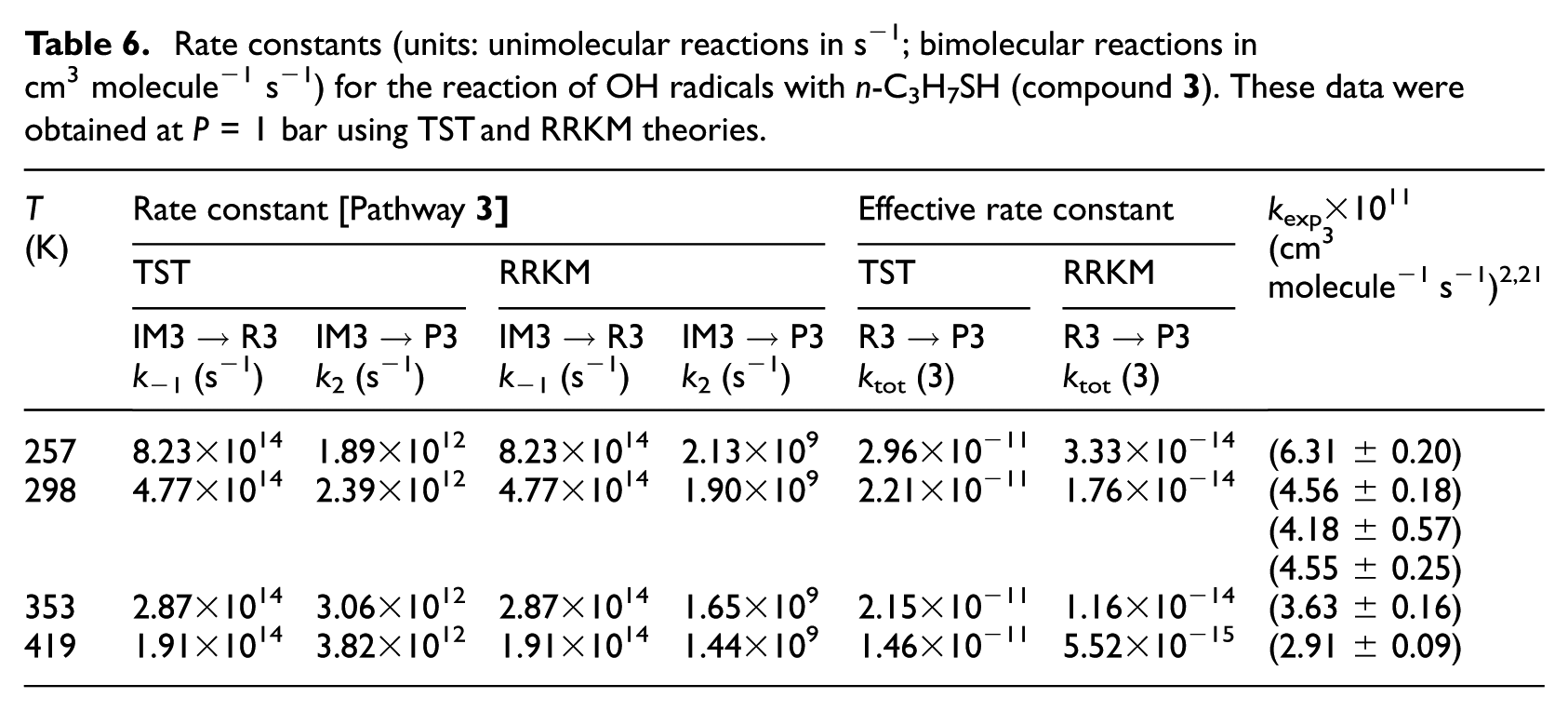
TST and RRKM estimates for individual rate constants of the studied compounds were calculated in the gas-phase and at a pressure of 1 bar and over the considered temperatures in line with the original experiments2,21 which are listed in Tables 4−7 and compared with experimental data. Furthermore, RRKM data computed at lower and higher pressures are provided for the same temperatures in Table S4(a)−(i) of the ESI. Theoretical values obtained using the TST at a pressure of 1.0 bar do not differ by more than one order of magnitude from the experimental data.
Rate constants (units: unimolecular reactions in s−1; bimolecular reactions in cm3 molecule−1 s−1) for the reaction of OH radicals with C2H5SH (compound
Rate constants (units: unimolecular reactions in s−1; bimolecular reactions in cm3 molecule−1 s−1) for the reaction of OH radicals with n-C3H7SH (compound
Rate constants (units: unimolecular reactions in s−1; bimolecular reactions in cm3 molecule−1 s−1) for the reaction of OH radicals with iso-C3H7SH (compound
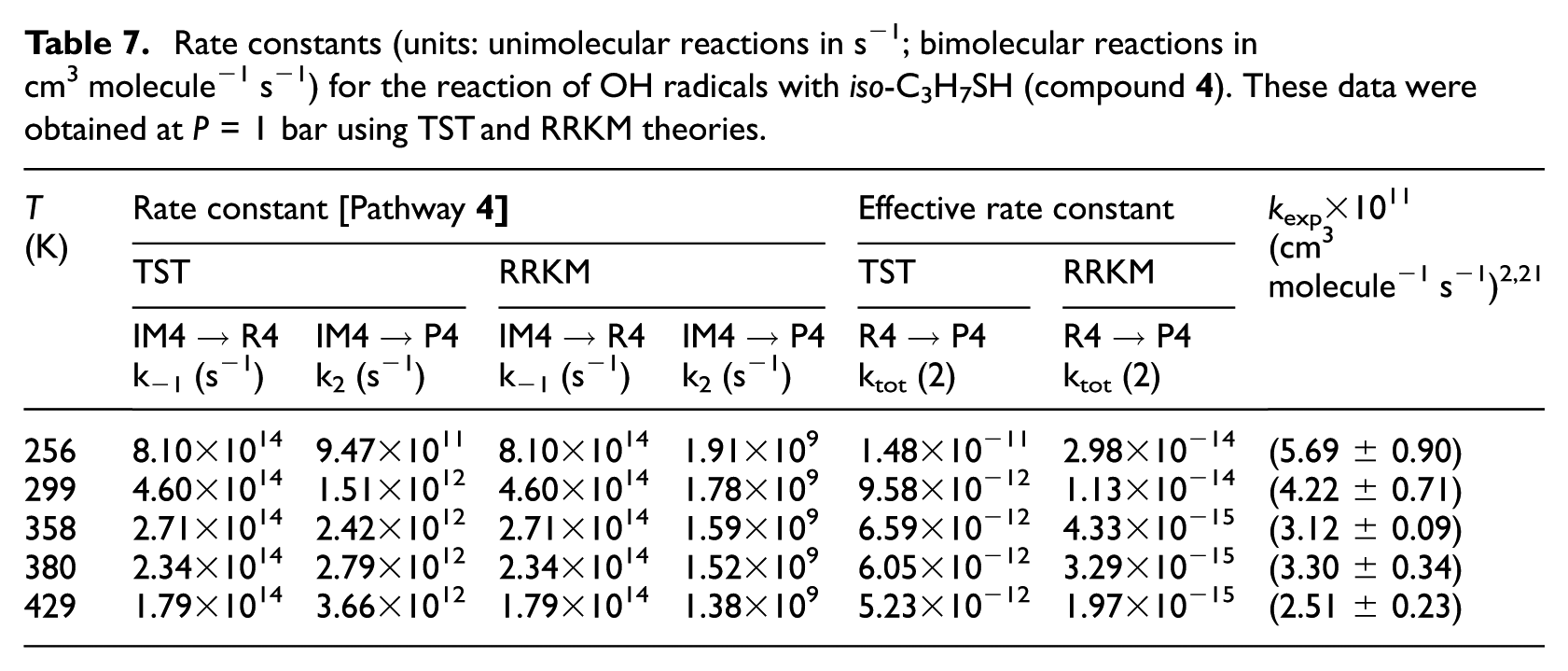
Effective rate constants for pathways
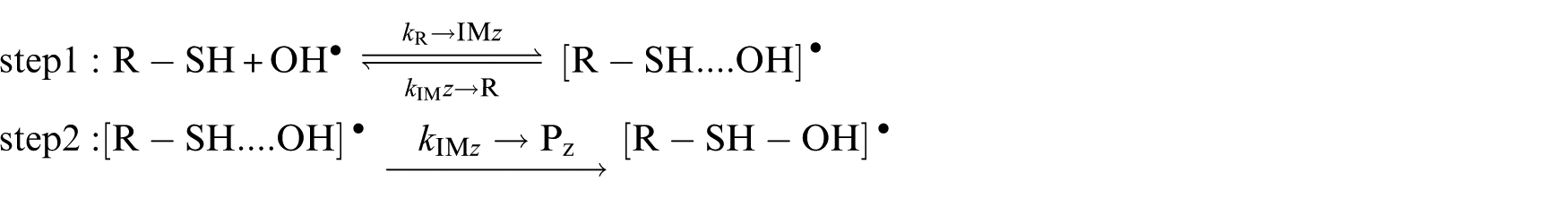
A steady state analysis of the above sequence of reactions leads to the following easily tractable expressions for the effective kinetics characterizing the studied chemical pathways:
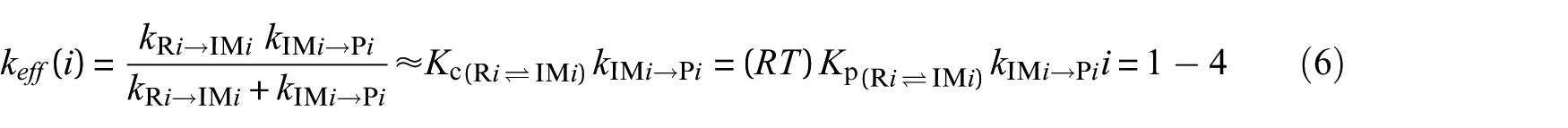
k[R→IMz,(z = 1−4)] represent the kinetics characterizing the forward bimolecular reaction step (in cm3 molecule−1 s−1), whereas kIMz→P and kIMz→R are the forward and backward unimolecular kinetics (in s−1), respectively. RRKM estimates for unimolecular (k2) and effective bimolecular kinetics (keff) are listed in Tables 4−7 (at P = 1 bar) and Tables 8 and 9 (at P = 75 Torr). These data were obtained from the computed CBS-QB3 energy profiles and B3LYP/6-311G(2d,d,p) microcanonical rovibrational densities of states. In these tables, theoretical kinetics can also be compared with available experimental data.2,21
Kinetic and effective rate constants for the OH-addition pathways of compounds
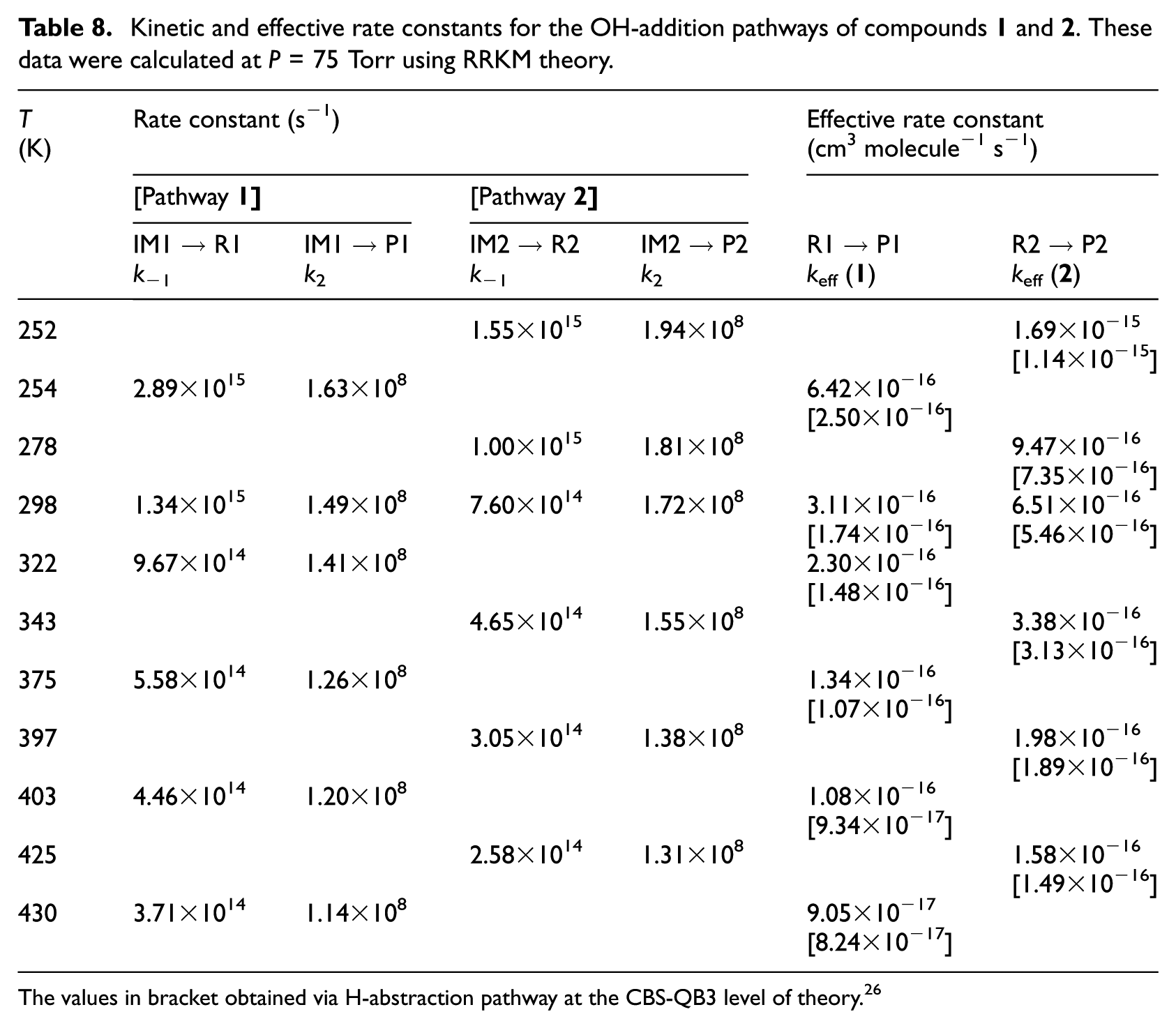
The values in bracket obtained via H-abstraction pathway at the CBS-QB3 level of theory. 26
Kinetic and effective rate constants for the OH-addition pathways of compounds
The values in bracket obtained via H-abstraction pathway at the CBS-QB3 level of theory. 26
Kinetic rate constants are the results of TST and RRKM calculations performed upon the CBS-QB3 energy barriers and densities of states. Further, RRKM data obtained at lower and higher pressures are given at the same temperatures in Table S4(a)−(i) of the ESI. The reader is referred further to Table S4(a)−(i) of the ESI for a detailed evaluation of the temperature dependence of the equilibrium constants (Kp) characterizing the first reversible reaction step. In line with the computed energy profiles and rate constants, the formation of the
An Arrhenius plot (Figure 3) of the effective rate constants which were obtained using RRKM theory for the chemical reactions
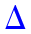
Arrhenius plot of the obtained RRKM bimolecular rate constants for the reactions of OH radicals with the studied aliphatic thiols using the CBS-QB3 approach (P = 75 Torr). Symbols:
As is to be expected, because of the involved negative energy barriers at the experimental pressure of 75 Torr, 21 RRKM effective rate constants obtained from the CBS-QB3 energy for the fastest reaction pathway (R3→P3) overestimate the experimental ones by ∼4 orders of magnitude at the investigated temperatures. In view of the known performances of the CBS-QB3 approach, this semi-quantitative agreement between theory and experimental is quite satisfactory, considering that hindered rotations of the OH group and anharmonic effects in the intermediate and transition states have been neglected. Furthermore, it appears quite clearly that pressures larger than ∼104 bar are required for ensuring a saturation of the computed bimolecular rate constants compared with the high-pressure limit (TST) of the RRKM bimolecular rate constants (see Figure 4).
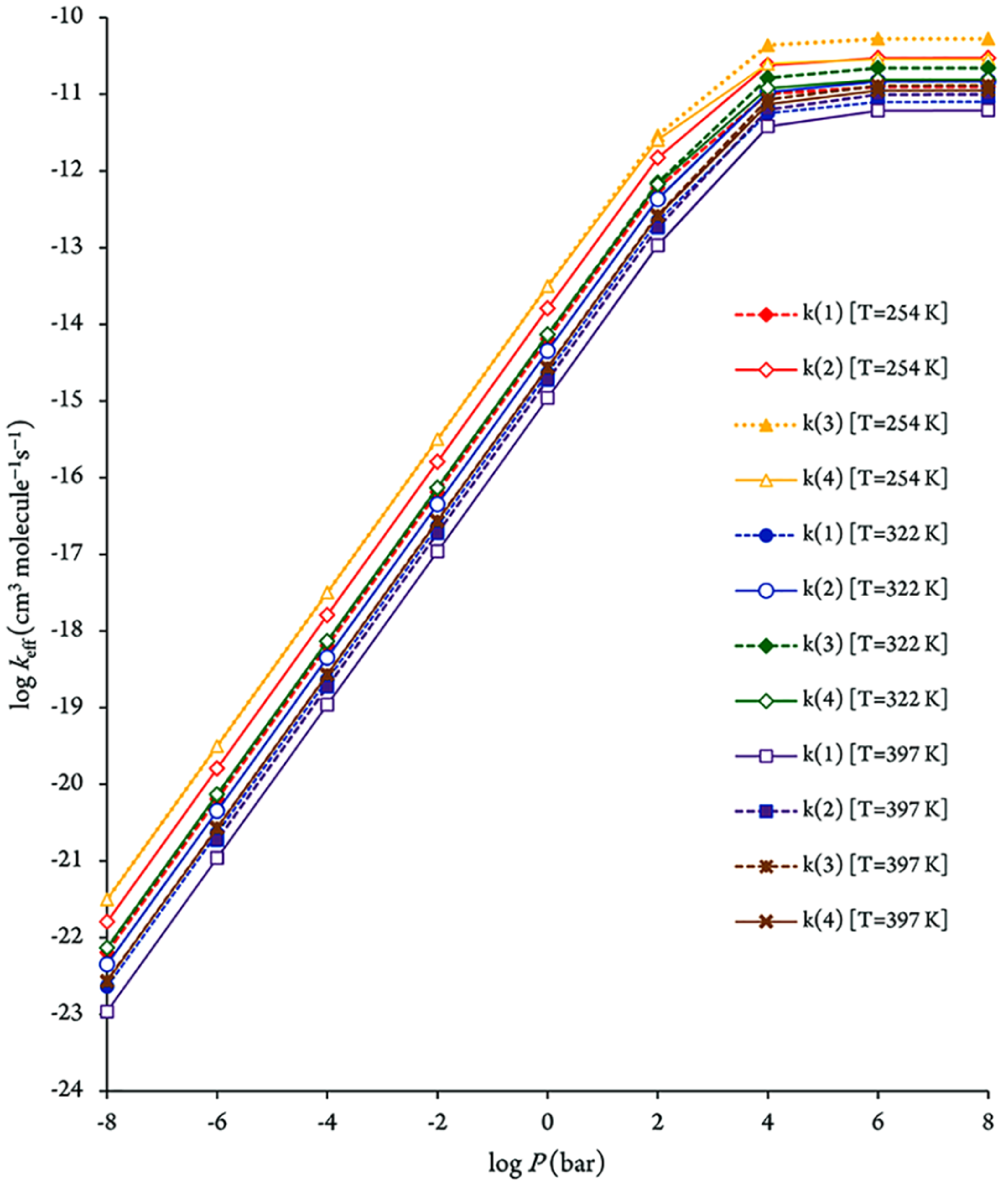
Plot of the bimolecular rate constants for R i →P i (i = 1−4) reaction pathways at a pressure of 75 Torr and the studied temperatures.
Consequently, the comparison with the RRKM data, nevertheless, indicates that the TST approximation breaks down at pressures higher than 104 bar for the kinetic rate constant characterizing pathway
The pressure dependence of the computed RRKM rate constants is depicted at 254, 322 and 397 K in Figure 4. From this figure, it is immediately apparent that rather high pressures (>104 bar) are large enough to ensure a saturation of the RRKM rate constants, in comparison with the high-pressure limit (TST). This observation is most obviously the consequence of the negative activation energies, which makes standard TST invalid at atmospheric pressure (1 bar). Pressure effects need, therefore, to be taken into account on RRKM grounds for consistent insights (Tables 8 and 9) into the experimental rate constants,2,21 which were obtained at a pressure of 75 Torr. Compared with the high pressure limit, the pressure effects result, indeed, in this case into a most significant decrease of the computed rate constants, by ∼4 orders of magnitude.
As a result of the differences in energy barriers between the OH-addition and H-abstraction reaction pathways (Table 3), and also lower barrier height levels for the OH attack onto sulphur atom in the studied compounds
Conclusion
The reactions between alkyl-substituted aliphatic thiol and hydroxyl radicals in the gas phase have been studied for the first time on computational grounds using at the composite CBS-QB3 level of theory. Kinetic rate constants for unimolecular and bimolecular reaction steps were estimated by means of TST and statistical RRKM theory.
This first reaction steps for OH addition via pathways
Effective rate coefficients have been calculated according to a steady state analysis upon a two-step model reaction mechanism, assuming reversibility of the first bimolecular addition reaction step and irreversibility of the second unimolecular dissociation step. In line with the experimental observations,2,21 this indicates that the kinetically most efficient process at temperatures ranging from 252 to 430 K corresponds to OH addition to n-C3H7SH. The results show that the kinetic rate constants decrease with increasing temperatures and decreasing pressure.
In line with negative activation energies, it was found that the standard transition-state-approximation breaks down at ambient pressure (1 bar) for the first bimolecular reaction steps. RRKM calculations show in particular that overwhelmingly high pressures, larger than 104 bar, are required for restoring of this approximation for all reaction channels involved in the oxidation of the studied pathways, in particular for the conversion of the pre-reactive vdW complex [R−SH…OH]• into the molecular energized adduct [R-SH−OH]•.
A comparison with TST results seems to validate RRKM theory for all studied pathways. RRKM theory appears to be sufficient for achieving semi-quantitative insights into the experimentally kinetic rate constants, with discrepancies in about ∼4 orders of magnitude between theory and experiment.
The supplied data indicate that, from a kinetic viewpoint, the most favourable process is reaction between n-C3H7SH and hydroxyl radicals through OH-addition pathway into the
Supplemental Material
PRK1800760_Supp_ – Supplemental material for Oxidation reaction mechanism and kinetics between OH radicals and alkyl-substituted aliphatic thiols: OH-addition pathways
Supplemental material, PRK1800760_Supp_ for Oxidation reaction mechanism and kinetics between OH radicals and alkyl-substituted aliphatic thiols: OH-addition pathways by Arezoo Tahan and Abolfazl Shiroudi in Progress in Reaction Kinetics and Mechanism
Footnotes
Acknowledgements
This paper has been derived from the research project entitled ‘Theoretical study on mechanisms and kinetics of the gas phase reactions of the hydroxyl radicals with aliphatic thiols containing short chain alkyl groups (1 to 3 carbon atoms)’. The authors would like to thank Prof. M.S. Deleuze for helpful discussions during the course of this investigation.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship and/or publication of this article.
Funding
The author(s) received financial support for the research, authorship, and/or publication of this article: This work was financially supported by the Semnan Branch, Islamic Azad University through Grant No. 17752.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.