
Abstract
New 1H and 13C NMR 400 MHZ spectra of the 2-Nb cation under stable ion conditions, for example, in SbF5/SO2F2/SO2ClF, −80 oC, show besides the usual 1H NMR resonances at δ 4.93, 2.82, 1.85, the never before seen singlet, δ 9.63, and doublet, δ 2.97 (J2,6 = 16.6 Hz), ratio 1.00 : 1.07, proposed to be due to resonance-stabilized bridgehead 1-Nb cationic enantiomers in equilibrium with 2-Nb cation. The corresponding 13C proton-coupled NMR spectrum, −80 oC, has a C3,5,7 triplet, δ 30.45, J(CH) = 139.14 Hz, and C4 doublet, δ 37.7, J(CH) = 154.54 Hz. The C1,2,6 absorption, δ 91.04 is relatively broad, whereas previously, at −70 oC, it was a pentuplet. The 13C proton-decoupled spectrum at −80 oC shows the C4 doublet and C3,5,7 triplet collapsed to a singlet, but the C1,2,6 resonance is still broad. Analyses support the slowing exchange between resonance stabilized enantiomeric 2-cations at ≤ –159 oC. Some future studies are proposed.
Introduction
The cornerstones of the structure proof of the 2-norbornyl cation1,2 are its 1H and 13C NMR (nuclear magnetic resonance) spectroscopic studies.3–9 It is crucial to recognize that the interpretations of these NMR studies at ≤ –150 oC are not secure.10–14 For example, the proton- coupled and decoupled 13C NMR spectra are (1) significantly different at -150 oC and −159 oC, (2) an illustration of the proton-coupled 13C NMR spectrum at − 159 oC has not been published, and (3) the coupling constants of some absorptions in the 13C NMR proton-coupled spectrum at − 150 oC have been incorrectly analyzed.14 Also, the interpretations did not consider more recent evidence indicating 1) anchimeric assistance of 2-exo leaving group displacement by bridgehead hydrogen orbital 2) presence of a pair of stereospecific front-face C1,2,6 hydride shifts via the bridgehead C1 rather than the assumed nonstereospecific 6,2,1 hydride shift for which there is no evidence 3) possibility of C1 instead of C6 methylene bridging, and 4) σ -aromaticity 14
The crystal structure of the 2-norbornyl cation in solvated [C7H11]+ [Al2Br7]–. CH2CBr2 salt at 40 K has been published and reported to be traditional nonclassical (TNC) structure I. 15 These carbon assignments are based upon nonsettled NMR interpretations, that is, the bridging methylene carbon is represented to be assumed C6, rather than C1. Mean bond distances were assigned as C1–C2, 1.39 Å and C1C6=C2C6, 1.80 Å rather than C2C6 being 1.39 Å, having double bond character, and C1C6=C1C2, structure II. The possibility that the crystal structure may have C-H lengthened bonds, for example, structure III, was not considered.
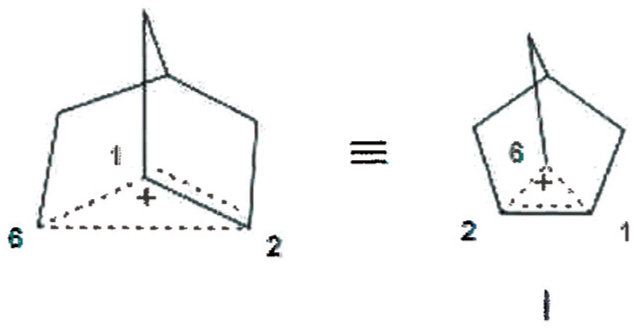
Traditional nonclassical 2-Nb cation with assumed C6 as bridging carbon in crystal structure.
Proposed alternative nonclassical 2-Nb cation with C1 as bridging carbon in crystal structure.

C-H lengthened bonds in nonclassical 2-Nb cation crystal structure was not mentioned as possibility.
Nevertheless, even though the crystal structure of the 2-Nb cation is nonclassical, does not answer the questions of its (1) structure in low nucleophilicity stable ion conditions, (2) original contentious questions of its transition states and intermediates in solvolyses of norbornyl derivatives, and (3) the high exo/endo rate ratios in solvolyses of its derivatives.
Presented here are new 1H, 13C proton-coupled and proton-decoupled NMR spectra and quenching experimental data, which indicate that a resonance stabilized form of the bridgehead 1-Nb cation may be in equilibrium with the 2-Nb cation under stable ion conditions at −80 oC. This supports the hypotheses that (1) the C1 bridgehead hydrogen orbital is involved in anchimerically assisting the displacement of the 2-exo-Nb leaving group, (2) the experimentally observed stereospecific hydride shifts in 2-Nb cation 16 proceed via a pair of equatorial hydride shifts, H1 to exo-C2 followed by exo-H6 to C114 rather than the nonstereospecific 6,2,1 hydride shift, which would result in scrambling of hydrogens, and (3) the bridging methylene carbon is C1 rather than C6 in the nonclassical 2-Nb cation. 14
Experimental*
Experimental materials, method, and apparatus
The 2-Nb cation was prepared from 98% exo-2-norbornyl chloride (Aldrich). A gas chromatogram showed no evidence of impurities and a mass spectrum showed ≤ 1% of 2-norbornanol as an impurity. Since this compound would also only yield the same intermediate 2-Nb cation as 2-NbCl, it was used as is, at ∼−78 ○C (dry ice-acetone), utilizing the procedure of Olah et al. 6 Spectra were reproduced with different sample preparations under three different stable ion media, that is, SbF5:FSO3H (1:1)/SO2ClF with and without SO2F2 and in SbF5/SO2ClF/SO2F2. Spectra were run on a Bruker 400 MHZ spectrometer.
Quenching of the 2-Nb cation solution was done at −66 °C with aqueous methanol, extracted with ether, dried 6 and analyzed by mass spectroscopy, products sorted by retention time, product percentages determined electronically.
Results
1H NMR spectra
Figures 1(a)–(c) are the previously unobserved 1H NMR spectra of the 2-Nb cation in SbF5/SO2ClF/SO2F2 at −80 oC, −119.7 oC, and −147 oC, respectively. In Figure 1(a), besides the usual previously observed peaks at δ 4.93 (4.0000 H), 2.82 (1.0066 H), and 1.85 (6.0103 H), there is the additional singlet at δ 9.63 and doublet at δ 2.97 (J2,6 = 16.6 Hz). The area ratio of singlet : doublet is 1.00 : 1.07. Lowering the temperature to −119.7 oC, Figure 1(b), results in a disappearance of the δ 9.63 peak and the δ 2.97 doublet is not seen. At −147.7 oC, Figure 1(c), the spectrum matches the previously published spectrum at −154 oC, Figure 1(d). 4 The original δ 4.93 peak has decoalesced into the downfield δ 6.75 and upfield δ 3.17 peaks. The possibility the δ 2.97 doublet has not disappeared and may be buried underneath the broad decoalescing peak at δ 3 peak cannot be ruled out. The electronic integration ratio read-outs of peaks at δ 4.93 (4H, 2-Nb) and δ 2.97 (2H, 1-Nb) are 4.0000 : 0.2007. Assuming equal peak areas/proton, means the doublet represents about 9.6% of the product. This suggests the doublet is an actual intermediate, not an impurity, break-down product or artifact. As discussed below, this is supported by the 13C NMR spectra. Previous integration analyses of the 1H NMR peaks at δ 4.93 (4H), 2.82 (1H), and 1.85 (6H) were accurate to one significant figure.
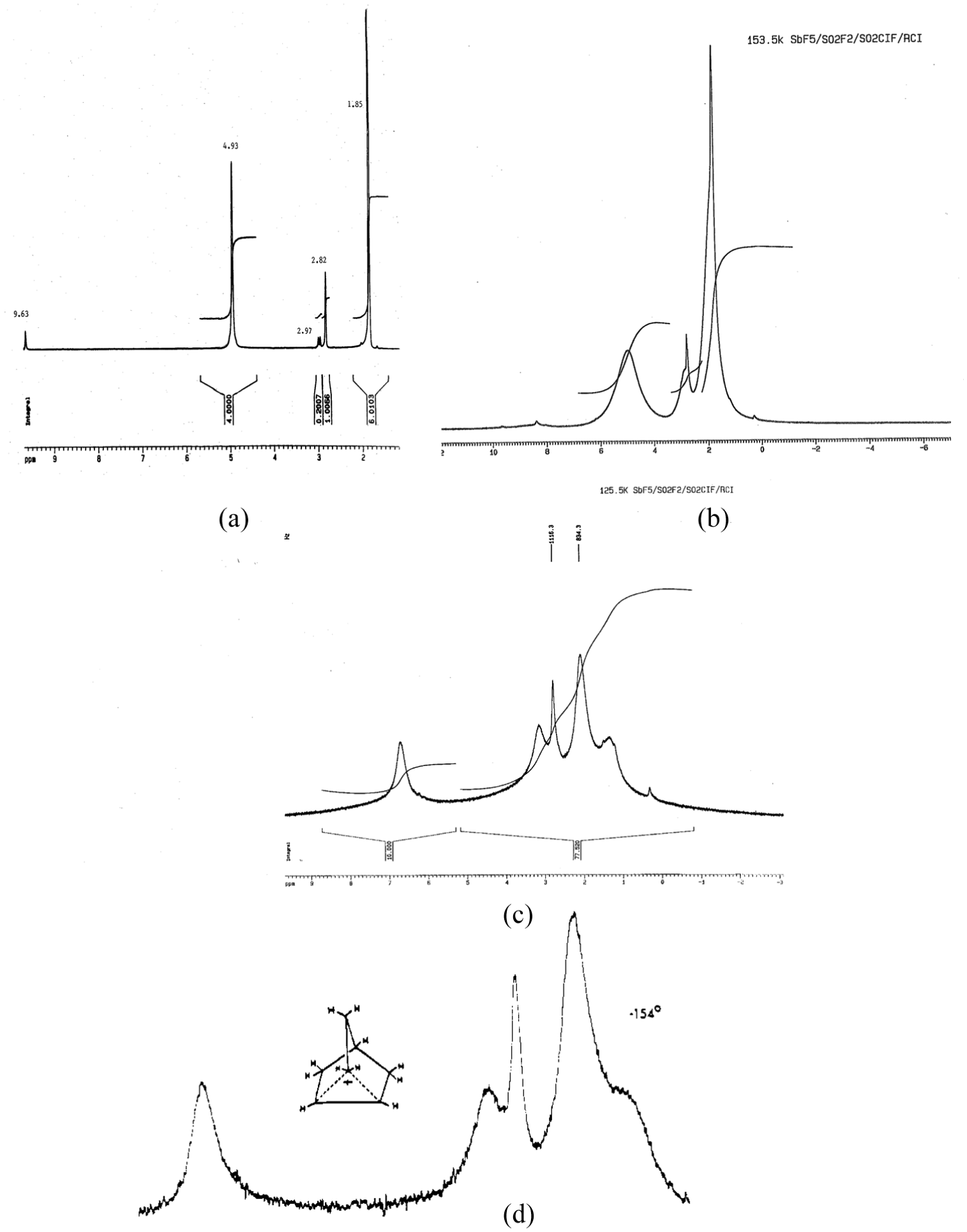
1H NMR spectra of 2-Nb cation, 400 MHZ, SbF5/SO2ClF/SO2F2 (a) −80 oC, (b) −119.7 oC, (c) −147 oC, (d) −154 oC, previously published, 100 MHZ (Source. Reproduced with permission from Olah and White. 4 ).
13C proton-coupled and proton-decoupled NMR spectra
The 13C proton-coupled spectrum at −80 oC, Figure 2(a), has a relatively broad low-field C1,2,6 absorption at δ 91.04, C4 doublet, δ 37.7, J(CH) = 154.54 Hz, C3,5,7 triplet, δ 30.45, J(CH) = 139.14 Hz. Figure 2(b) is the corresponding 13C proton-decoupled spectrum at −80 oC, and for comparison purposes, the previously published Fourier transform 13C, off-resonance spectrum, −70 oC, is Figure 2(c). 7
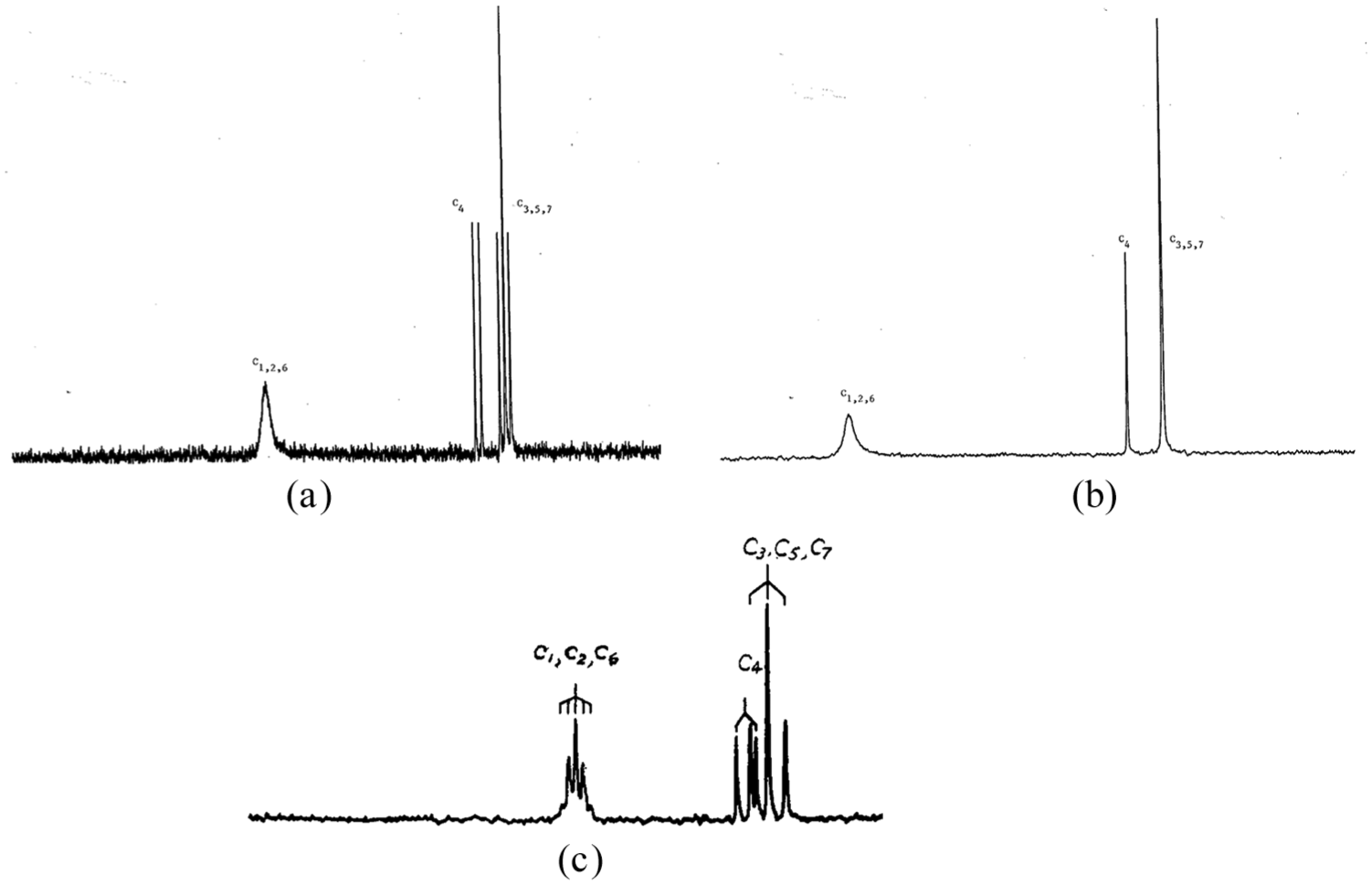
13C NMR spectra of 2-Nb cation, 400 MHZ, SbF5/SO2ClF/SO2F2, −80 oC (a) proton-coupled, (b) proton-decoupled, (c) Fourier transform, off-resonance, −70 oC (Source. Previously published, reproduced with permission from Olah et al. 7 ).
Quenching
Quenching the 2-Nb cation solution at −66 oC with aqueous methanol yielded 37.7% 1-hydroxynorbornane, 4.2% 1-chloronorbornane, 0.9% 1-methoxynorbornane, 43.5% dinorbornylether, 0.4% 2-hydroxynorbornane, and 13.3% unidentified products.
Discussion
1H NMR spectra
It is proposed that the singlet at δ 9.63 and doublet at δ 2.97 (J2,6 = 16.6 Hz), with area ratio 1.00 : 1.07 may be due to the presence of a stabilized form of the bridgehead 1-Nb cation, Scheme 1(b), which is in equilibrium with the 2-Nb cation, which can undergo Wagner–Meerwein shifts and a pair of exo and bridgehead hydride shifts via the bridgehead C1, Scheme l(a). 14 It is doubtful if the nonstabilized 1-Nb cation is present.
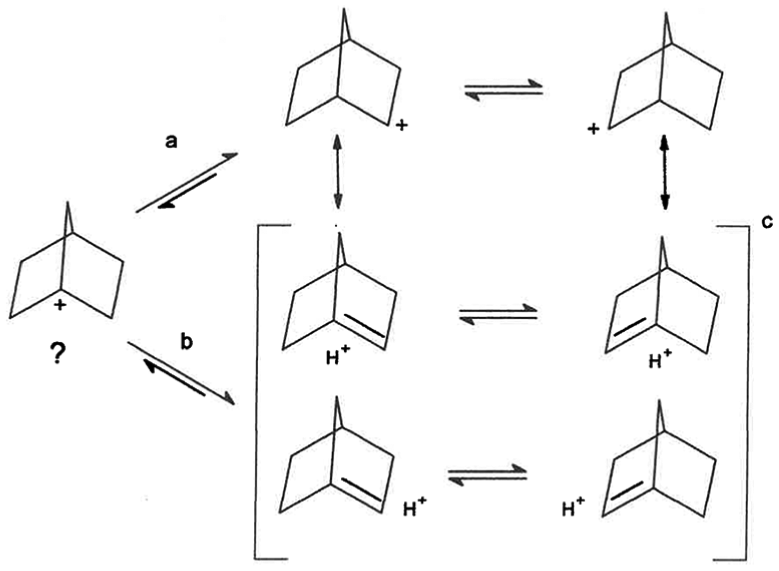
(a) Proposed equilibrium between 1-Nb cation and 2-Nb cation, SbF5/SO2ClF/SO2F2, −80 oC, (b) anti-Bredt olefin 1-Nb cation enantiomers in equilibrium, (c) matching carbon frameworks of 1-Nb cation and 2-Nb cation.
The low-field δ 9.63 peak may be due to the two exo protons, which can stabilize the 1-cation by hyperconjugation, and the δ 2.97 doublet may be due to the two endo protons coupled to each other, but not coupled to the longer, weaker hyperconjugating exo protons, Figure 3,
In the allyl cation
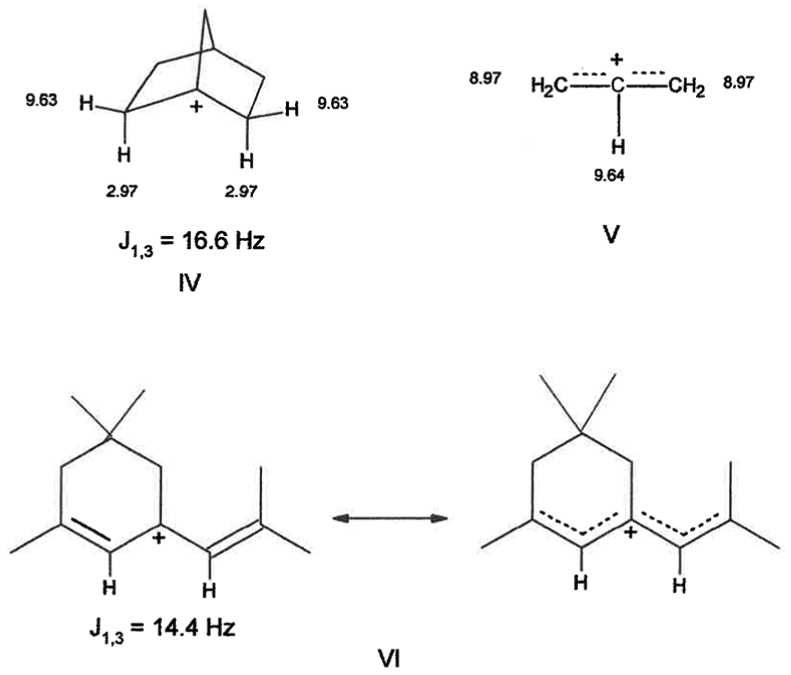
Also supportive of the above interpretation is the disappearance of the δ 9.63 peak upon lowering the temperature, which yields a spectrum, Figure 1(c), which matches that published at −154 oC, Figure 1(d). 4 However, the possibility the δ 2.97 doublet has not disappeared and may be buried underneath the broad decoalescing peak at δ 3 peak cannot be ruled out. The electronic integration analysis indicates the doublet represents 9.6% of the product suggesting that it is an actual intermediate.
A rationale for the observation of only two new 1H NMR peaks, which are assigned to the 1-Nb cation, is the following: The 1- and 2-Nb cations have resonance-stabilized hyperconjugated structures whose carbon skeletal frameworks are basically identical, for example, cf., Scheme 1(c). The stand-out difference is the position of H. If the new peaks in the 1H NMR were due to an impurity or break-down product, some other resonances would be expected in the corresponding 13C NMR at −80 oC, but there were none. Interestingly, the 13C NMR absorption of ethylene, δ 124.5 in SO2ClF, −120 oC, 20 is same as the base carbons of 2-cation at −159 oC, even though it has quite a different skeletal structure. Olah et al. have stated “For all practical purposes the unsymmetrical bridged ions 1b would be indistinguishable from the symmetrically bridged system 1a.” 8
Other possible explanations for the δ 2.97 doublet, for example, it may be due to coupling between the endo 6-H and endo 2-H and their corresponding anti 7-H, which are in the W-configuration or gem coupling with the exo-Hs, would require explanations for lack of absorptions of 7-Hs and gem exo-Hs, respectively.
13C proton-coupled and proton-decoupled NMR spectra
In the new 13C proton-coupled NMR spectrum at −80 oC (Figure 2(a)), the C3,5,7 triplet at δ 30.45, J(CH) = 139.14 Hz is well separated from the C4 doublet at δ 37.7, J(CH) = 154.54 Hz in contrast to the published Fourier transform, off-resonance spectrum at −70 oC, Figure 2(c). 7 In the low-field region of new spectrum, the C1,2,6 absorption at δ 91.04 is relatively broad and not split, whereas at −70 oC, this absorption is a pentuplet. The latter is attributed to a fast equilibrium due to “three equivalent carbons associated with four equivalent protons assigned to the cyclopropane(like) ring.” 3 This comparison of low-field results, in conjunction with the seeming anomaly between the broad nonresolved δ 91.04 absorption, whereas the high-field region is well-resolved at −80 oC, indicates something other is occurring besides a fast equilibrium due to “front-face” C1,2,6 carbons associated with equivalent protons. The broad nonresolved absorption may be due to the presence of resonance stabilized 1-Nb enantiomeric cations, which have a very similar skeletal framework as the 2-Nb cation, Scheme 1(c).
Additional support for this is the following. In the 13C NMR spectrum of 2-Nb cation, even at −159 oC, there is 2 to 2.5 ppm line broadening attributed to slow exchange of 6, 2, 1 hydride shift. 21 A pair of stereoelectronically aligned front-face “equatorial” hydride shifts via bridgehead C1 yields the enantiomer, Scheme 1(a). The slowing of the pair of hydride shifts would result in a slowing of exchange between enantiomeric 2-Nb cations, could result in observed line broadening. Also, in solid-state studies, even though there is no change in the δ 125 carbon absorption between −144 oC and −196 oC, there is broadening of all resonances. 22 Kramer and Scouten 11 have analyzed these published spectra and concluded there is selective line broadening of the peak at δ 125. They suggested it may be due to slowing of postulated exchange between enantiomeric Nb cations. The researchers of the solid-state studies confirmed there is indeed a selective line broadening of the resonance at δ 125. 23 Suggested here, the selective line broadening could be due to a slowing of a pair of front-face “equatorial” exo and C1 hydride shifts, that is, C1 to C2/C6 to C1, and/or slowing of Wagner–Meerwein shifts resulting in a slowing of exchange between enantiomeric 2-Nb cations. This is supported by C-14 labeling studies, which postulate the presence of more than a single cationic intermediate, whether it is classical or nonclassical.24–27 The enantiomeric pair of cations may be classical, Scheme 1(a), or unsymmetrically bridged nonclassical cations, equation (1).
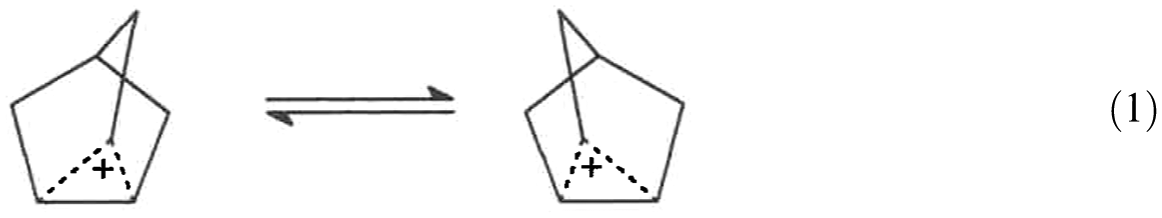
It is important to recognize that the 6,2 “1,3-type” hydride shift was proposed, without any evidence, to explain the C-14 label at C5,6 because no combination of 1,2-alkyl or hydride shifts could explain the results. However, hydride shifts via bridgehead C1 were excluded from consideration. 24 Accordingly, the equilibration does not have to proceed via a symmetrically C–C σ-bridged structure as previously suggested, 9 and Scheme 1(a) or equation (1) may be operative.
The suggestion of an equilibrium between 1- and 2-Nb cations is supported by the corresponding 13C proton-decoupled spectrum at −80 oC (Figure 2(b)), which shows that the C4 doublet and C3, 5, 7 triplet peaks collapse to a singlet, whereas the C1,2,6 peak is still broad and not resolved. On the other hand, in the previous corresponding 13C NMR spectrum at −70 oC, Figure 2(c), all the peaks collapsed to a singlet upon proton-decoupling supportive of the presence of just the 2-cation isomer. The latter result, once again contrary to the new spectrum, indicates C1,2,6 are equivalent.
Quenching solution of cation
The presence of the 1-Nb cation in equilibrium with the 2-Nb cation at −80 oC is strongly supported by quenching the solution at −66 oC with aqueous methanol, which yielded 42.8% of 1-substituted products (37.7% 1-hydroxynorbornane, 4.2% 1-chloronorbornane, 0.9% 1-methoxynorbornane.) Previous quenching of 1-chloronorbornane in SbF5-SO2 at −70 oC, after warming at −50 oC, −35 oC, and −20 oC yielded mixtures of 1-norbornanol and exo-2-norbornanol. 28 The authors state “the simplest conceptual mechanism for the transformation of IV (1-chloronorbornane) to the 2-cation is a simple 1,2-hydride shift.” If the hydride migration was intermolecular, there would have been a significant yield of chloronorbornanol in these solvolyses, but there was none. The hydride migration is intramolecular and by microscopic reversibility principle, the C1 to C2 hydride migration is intramolecular. Furthermore, the antimony pentafluoride donor–acceptor complex with 1-NbCl slowly rearranges to the 2-cation and attempts to observe the 1-cation were unsuccessful. 29 Unfortunately, only the 13C NMR is available, the 1H NMR is not. The very large antimony pentafluoride complex blocks the face of the incipient 1-cation sterically hindering an intermolecular hydride migration. Hence, the formation of 2-Nb cation means, once again, that the hydride migration is intramolecular. All the hydride migrations in mono-substituted norbornyl systems and related bicyclic structures are intramolecular.30–32 These results strongly support the hypothesis of pair of front-face “equatorial” hydride shifts via the bridgehead position rather than the nonstereospecific 6,2,1 shift for which there is no evidence and has been assumed.
Conclusion
NMR studies, quenching results
The singlet, δ 9.63 (2H), and doublet, δ 2.97 (2H), in the 1H NMR −80 oC may be the exo and endo absorptions, respectively, of the resonance stabilized 1-Nb cation enantiomers in equilibrium with the 2-Nb cation enantiomers. This is supported by the broad low-field absorption at δ 91.04 in the corresponding 13C NMR, both proton-coupled and decoupled, which is in contrast to the well-resolved upfield region of the proton-coupled spectrum. In solution at −159 oC, there is a 2–2.5 ppm line broadening, 21 and in solid-state studies between −144 and −196 oC, 22 there is confirmed selective line broadening of the δ 125 carbon absorption 23 suggested to be due to slow exchange between enantiomeric Nb cations. 11 These results complement and support our new results and analyses, which are in agreement that there is a slow exchange between enantiomeric cations, and postulate that it proceeds via a slowing of a pair of front-face “equatorial” hydride shifts through bridgehead C1 and/or slowing of Wagner–Meerwein shifts. The equilibration does not have to proceed via a symmetrically C–C σ-bridged structure as previously suggested. 9 The enantiomeric pair of cations may be classical, Scheme 1(a), or unsymmetrically bridged nonclassical cations, equation (1).
The quenching of 2-Nb cation solution with aqueous MeOH, which yielded 42.8% of 1-substituted products indicates resonance stabilized 1-Nb cation is indeed present in solution along with the 2-Nb cation. These results complement previous quenching results of 1-chloronorbornane in SbF5-SO2 at −70 oC which, for example, after warming to −50 oC yielded 65% of 1-norbornanol and 0.5% exo-2-norbornanol. 28
Formation of 2-Nb cation from β-Δ3-cyclopentenylethyl halides and 2-norbornyl halides
The 2-Nb cation can be prepared in various ways, for example, from β-Δ3-cyclopentenyl halides, structure
Its formation from
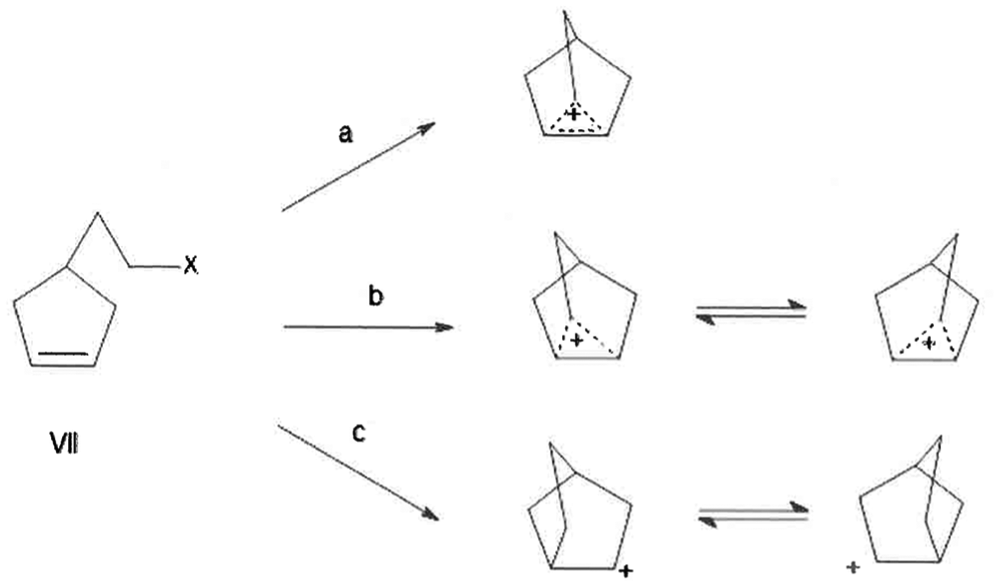
Anchimeric assistance by double bond in
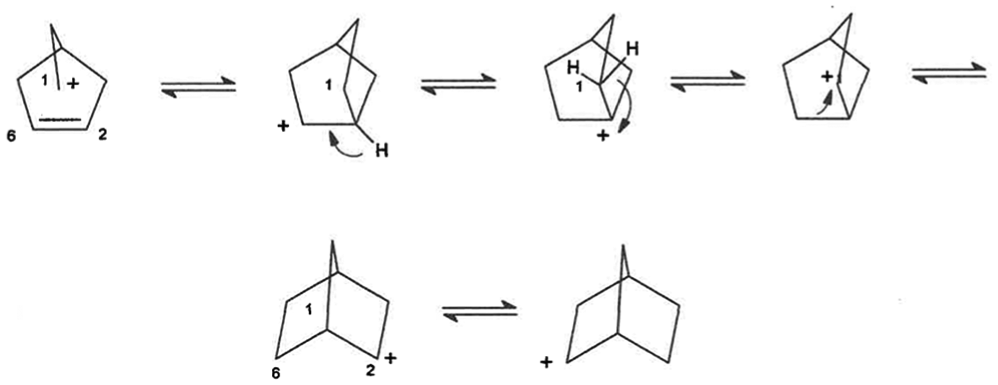
Representation of rearrangment of
For simplicity of visualization, the rearrangement is shown as proceeding via classical cations even though nonclassical cations are also a possibility. In formation of the nonclassical cations, in its simplest conceptualization, C1 may be the bridging carbon without breaking any C–C single bonds. The proposed mechanism in Scheme 3, involving a pair of hydride shifts via bridgehead carbon, is analogous to that proposed for ionic protoadamantane intermediate forming adamantane. 14
Previously missed observations
The dihedral angle between the Nb bridgehead C1-H orbital and C2–Cl exo orbital is 40.3o, whereas the dihedral angle between the C1-H and C2-Cl endo orbital is 72.0o. 14 The combination of significant differences in electronic environments, norbornyl ring strain and σ-aromaticity help enable the bridgehead C1-H orbital to anchimerically assist the displacement of the C1 exo leaving group in solvolyses of 2-exo substituted norbornyl derivatives.
The norbornane framework may be viewed as exhibiting σ-aromaticity, a saturated counterpart of a conjugated π system, for example, benzene. 14 The equatorial belt of overlapping exo orbitals with somewhat overlapping bridgehead bond orbitals in norbornane creates a closed circuit electron cloud by which through-space inductive effects preferentially stabilizes an equatorial exo transition state (TS). In high resolution NMR of saturated six-membered rings, there is a consistent tendency for signals of equatorial protons to appear at lower field than axial protons. It has been suggested this could be due to a ring current, which might be about one-third the ring current in benzene. 36
The new σ-aromaticity concept may account for the preference of exo products in norbornane substitutions, exo additions to norbornene, 1 and help explain preferential endo rather than exo stereoselectivity in Diels–Alder (DA) reactions.37,38 The steric interactions in the DA endo TS are not only more favorable when the incipient bridgehead protons are syn to exo C2 protons rather than the larger portion of the molecule in the exo TS, but also there are electronic stabilization effects in the endo TS not present in the exo TS, Scheme 4.37,38 Reviews of proposed DA mechanisms have been presented.39–43
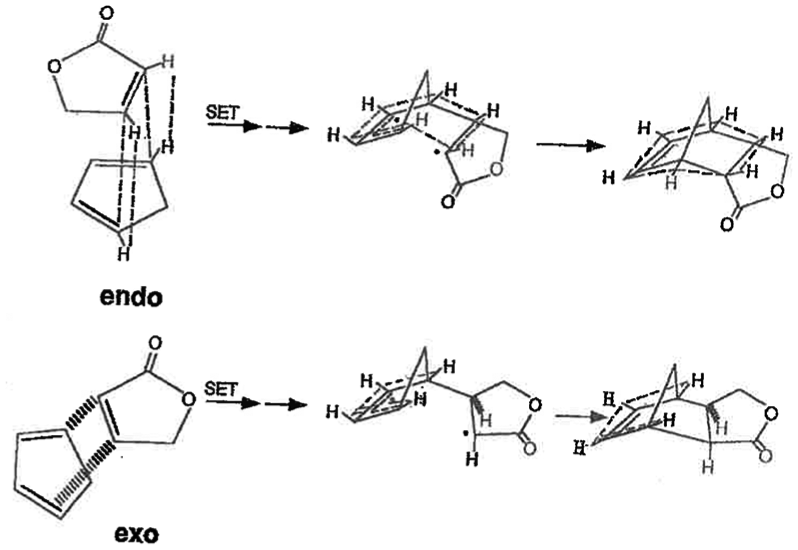
Representation of Diels–Alder exo and endo addition. Electronic stabilization effects, for example, σ-aromaticity, greater in endo transition state.
Correspondence between Brown, Dewar, and Olah
The core electron spectroscopy (ESCA) studies have been debated. There is no consensus as to the intensity ratio of the two bands, that is, whether the ratio is 6 : 1, as per Brown 44 and Dewar, 45 which would support a classical cation, or ∼ 5 : 2 as per Olah, 46 a nonclasssical cation. On the other hand, both Brown and Dewar state the 2-cation is unsymmetrical and Olah, whose correspondence is titled “Agreeing with Dewar,” seems to agree.
Lack of detection of stabilization energy
H.C. Brown’s insistence of an answer for the lack of detection of nonclassical stabilization energy in the exo TS in either the transition states for secondary exo-Nb derivatives (6.0 kcal) not present in the corresponding exo tertiary derivatives, or in the free secondary cation (∼ 8.0 kcal) not present in the tertiary cation has played a critical role in keeping the problem ongoing. At his ESOC V Conference lecture on the 2-Nb cation (Brown seems to have publicly withdrawn from subject after this conference), there was no offer of an explanation from the audience to his directly asked question. 47 The sole response, from one audience member, “It’s a shame Saul Winstein isn’t here.” More recently, an answer to Brown’s question of lack of detection of stabilization energy is that the bridgehead hydrogen orbital assists the displacement of the exo-C2 leaving group in both secondary and tertiary systems. 14
Future studies
The 1H NMR resonance assignments of 2-Nb cation in peak decoalescing temperature range, −148 to −159 °C, are tenuous. Also, the proton- coupled and decoupled 13C NMR spectra are (1) significantly different at −150 oC and −159 oC (2) an illustration of the proton-coupled 13C NMR spectrum at − 159 oC has not been published, and (3) the coupling constants of some absorptions in the 13C NMR proton-coupled spectrum at − 150 oC have been incorrectly analyzed. 14 Hence, it would be useful to obtain 13C NMR of 2-Nb cation at −150 oC and −159 oC, both proton-coupled and decoupled, to clarify results. Also, modern two-dimensional 13C/1H NMR correlation studies can be performed, for example, COZY and HMQC in peak decoalescing temperature range to obtain 13C and 1H NMR resonance assignments and one-to-one correspondence for each bonded pair of carbon and hydrogen. 48
Since a pair of front-face “equatorial” hydride shifts are postulated and probably occurred in 2-Nb cation, it would be interesting to obtain 1H and 13C NMR of cationic structures such as
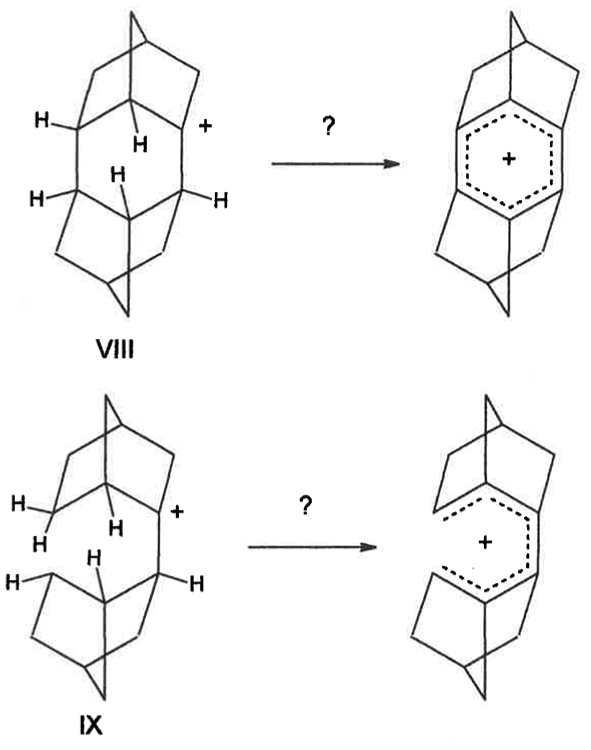
Suggested cationic structures
The 1-Nb cation can be generated in the gas phase, and in second mass spectroscopy experiment it may be isolated and fragmented. If it is stable, it will not isomerize to 2-Nb cation before it fragments. The results with analogous 2-Nb MS/MS experiment are compared. Performing the experiments in gas phase reduces the chance of bimolecular isomerization. 48
It has been proposed the 2-Nb cation can undergo an enormous number of degenerate rearrangements which is associated with high configurational entropy and unusual stability.11,49 This idea has been challenged50,51 and rebutted. 52
Spectroscopy studies of the 2-Nb cation, for example, ESCA, 1H and 13C NMR can be and have been challenged as to interpretation.10–14 On the other hand, it is undeniable that some form of the 1-Nb cation, resonance stabilized, is present in stable ion solutions of the 2-Nb cation as evidenced by quenching experiments of both 1-Nb 28 and, in this work, the 2-Nb cations. Also, labeling studies evidencing asymmetry, as demonstrated by uneven distribution of C13/C14 label, have not been challenged and seem impossible to refute.24–27
More research is needed. Despite various statements concerning its conclusion, the solution to the elusive 2-Nb cation has bedeviled researchers throughout its legendary six-decade existence.
Supplemental Material
Nb_Webbemail12_13_p2v51_l_ll_lllOKAM – Supplemental material for 1-Norbornyl cation may be in equilibrium with 2-norbornyl cation
Supplemental material, Nb_Webbemail12_13_p2v51_l_ll_lllOKAM for 1-Norbornyl cation may be in equilibrium with 2-norbornyl cation by Andrew Mamantov in Progress in Reaction Kinetics and Mechanism
Footnotes
Acknowledgements
The author gratefully acknowledges Professor Richard M. Pagni, Chemistry Department, University of Tennessee, for the use of his laboratory facilities; Dr George Hondrogiannis for the experimental work; Drs Jim Adcock and Isadore Sauers for the SO2F2; Dr Andrei Gakh for the experimental SO2F2 transfer; Drs Craig Barnes and Patti Kreke for help with the NMR instrumentation; and Dr Thomas H. Webb, Syracuse Research Corporation for the graphics. Also, a heartfelt thank you to these very kind people who performed the work gratis and for their time and efforts. The supplies, use of equipment, and laboratory were also gratis.
Declaration of conflicting interests
The author declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Dedication
This article is dedicated to Professor Herbert C. Brown and Dr. George M. Kramer.
Funding
The author received no financial support for the research, authorship, and/or publication of this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.