
Abstract
By employing a combined approach of the unity bond index–quadratic exponential potential method and density functional theory within the generalized gradient approximation, we have studied the interaction of intermediates in the ethanol dehydrogenation process to ethyl acetate on Cu, Ag, Ni, Pd, Pt, Co, Au and Ir(111) transition metal surfaces. Binding energies and geometries were optimized for the main intermediates of this process. Electronic structures were computed for some intermediates/transition metal systems. We also calculated the activation energies for the elementary steps of the reactions. The results show that amid the studied surfaces, Cu(111) stabilizes ethoxy and acetyl species, preventing their dissociation. Inducing the η2 binding mode of acetaldehyde by alloying Cu with Ni, Co, Pd, Pt or Ir can enhance the catalytic proprieties of the Cu(111) clean surface.
Keywords
Introduction
The worldwide demand for ethyl acetate (EA) has accelerated over recent decades by virtue of its broad application areas: agriculture, organic synthesis, adhesives, coatings and various other industries. It is essentially used as a solvent, the best alternative to supersede many conventional aromatic solvents. 1 Three industrial processes manufacture most EA. The first is esterification between ethanol and acetic acid, quickened by sulphuric acid under mild operating conditions. The second is by the disproportionation of acetaldehyde (Tishchenko reaction) in the presence of aluminium triethoxide; this reaction proceeds under atmospheric pressure and at low temperature (0 °C–5 °C). The third is based on the reaction between acetic acid and ethylene, using heteropolyacids like tungstosilicic acid dispersed on a support; typical operating parameters of this process are 77 bar and 150 °C.2,3 These three processes introduce serious technical disadvantages as well as economic and environmental issues. The use of liquid acids, either as catalysts or reagents, exacerbates corrosion. Homogeneous catalysts are not beneficial owing to difficult recovery from the reaction phase. An added shortcoming is related to the exothermic nature of the transformation, as in the case of Tishchenko reaction, for which intensive cooling is necessary. 2
In 2001, the Anglo-Norwegian engineering and construction group Kvaerner announced the commissioning of Sasol’s 50,000 tonnes/year EA plant in Secunda, South Africa. The technology employs the newest route to EA in which ethanol is dehydrogenated on a Cu/Cr2O3-based catalyst, under moderate operating conditions of 13–14 bar and 220–240 °C (equation (1)). The plant provides hydrogen with sufficient purity that it needs no additional processing. The aldehydes and ketones formed by secondary reactions are excluded in a selective polishing step, before the liquid phase is distilled to produce 99.5% min EA, and the residual ethanol is recycled to the dehydrogenation section. 4
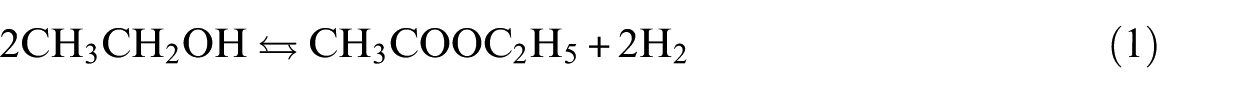
It is widely known that alcohols are easily dehydrogenated over solids containing copper to yield aldehydes, ketones and esters with distinct selectivities; typical heterogeneous catalysts are pure copper and copper dispersed on oxides such Cr2O3, ZrO2, ZnO and others. Previously, Iwasa and Takezawa studied the dehydrogenation of ethanol over Cu and Cu/(SiO2, ZrO2, Al2O3, MgO and ZnO) oxides. 5 They presumed that the reaction proceeds on reduced copper, thus ultraviolet (UV)/Vis spectra showed the presence of Cu(0) identified by an absorption edge at 560 nm for all active catalysts. However, the unreduced catalysts and supports were inactive. Colley et al. 6 confirmed the critical importance of Cu sites, using temperature-programmed reaction spectroscopy (TPRS); they found that ethanol (EtOH) first reacts with the Cu element of Cu/Cr2O3 catalysts to produce ethoxy (EtO), which in turn mutates into acetyl (MeCO). These two adsorbed species react and transform into an adsorbed EA molecule. However, other authors reported another mechanism in the case of Cu-Zn-Zr-Al-O multi-component catalysts.7,8 They proposed that throughout the process, acetaldehyde (MeCHO) is first generated from ethanol, which then unites to form hemiacetal and further dehydrogenated to EA. Theoretically, ethoxy molecules could combine to form diethyl peroxide and acetyl radicals combine to form diacetyl, as possible pathways. However, neither species was reported experimentally.6,7
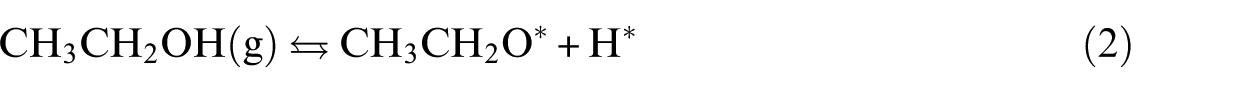


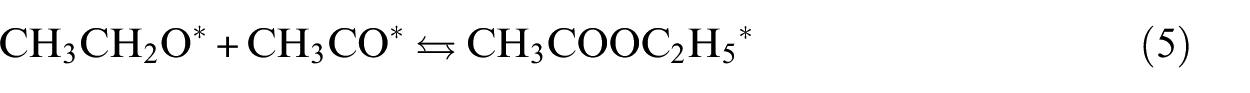
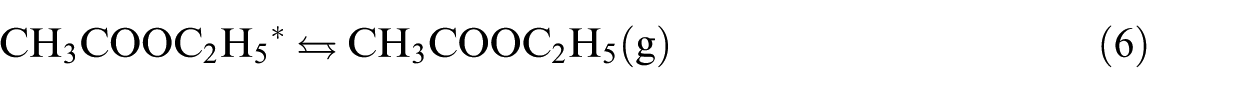
We aimed in this article to study the interaction of dominant intermediates in ethanol dehydrogenation to EA with Cu, Ag, Ni, Pd, Pt, Co, Au and Ir transition metal (TM) surfaces from a theoretical perspective, by using unity bond index–quadratic exponential potential (UBI-QEP) and density functional theory (DFT) approaches. Despite the intensive experimental works that highlighted the critical role of metallic copper in the dehydrogenation of ethanol, its mechanism of action has remained unclear. We believe that theoretical methods offer great opportunities for understanding microcatalytic phenomena, as well as identifying trends in the catalytic behaviour of metals in the foregoing reaction (equation (1)). Moreover, they could provide key insights into the principles for designing catalysts with enhanced properties.
Theoretical perspectives and computational details
All surface energetics calculations were based on two theoretical constitutions: DFT as implemented in Quantum ESPRESSO 9 and UBI-QEP. 10 For the first approach, we considered M(111) p (2 × 2) surfaces constituted of three layers of M atoms (M = Cu, Ag, Ni, Pd and Pt). The two lower layers were frozen in their positions to represent the bulk structure, while the top layer was let to relax. A vacuum width of 12 Å to separate slabs was required to eliminate any interactions induced by periodic boundary conditions (Supplemental material). The energies of the optimized structures were obtained by employing the generalized gradient approximation (GGA)–Perdew, Burke and Ernzerhof (PBE) functional. 11 Ultrasoft pseudopotentials (USPPs) 12 were chosen to describe electron–ion interactions, within a plane wave basis with energy cut-offs of 30 and 240 Ry for the wave function and charge density, respectively. Furthermore, a 4 × 4 × 1 k-point grid was used according to the Monkhorst–Pack method to effect the integration in the Brillouin zone. The binding energy of the intermediates was calculated using the following formula:

here, EA/S is the total energy of the M(111) surface with adsorbed molecules A, ES is the energy of the clean surface and EA represents the energy of A in the gas phase. Positive values of Eads indicate favourable adsorption energies. For low binding energies of 5–20 kcal mol−1, those of physisorption, van der Waals interactions are crucial. However, they are not well described by GGA-PBE that often tends to underestimate them, so we corrected Eads taking into account D3 dispersion. 13 For the case of EA physisorption, we added 4.6 kcal mol−1 (0.2 eV) to the value calculated by equation (7). Spin polarization was allowed for calculations with Ni.
The equations used to calculate adsorption energies, Eads, and activation energies, Eact, according to the UBI-QEP method were detailed intensely elsewhere by Shustorovich et al.10,14–19 In general, molecules can interact (1) weakly, (2) strongly or (3) moderately with the surface. Closed-shell molecules represent the first category for which the molecular electronic structure is barely perturbed after the adsorption (H2, CH3CH2OH, CH3CHO and EA). For a given diatomic or quasi-diatomic molecule AB that interacts weakly via its ‘A’ atom, the molecule’s binding energy

DAB is the enthalpy needed to break the bonds between the contact atom A and the rest of the atoms in fragment B. The strong interaction between the adsorbate and the substrate surface creates new types of chemical bond; this group includes molecules with unpaired electrons, which may have more than one unpaired electron, which occupies separate orbitals in its outer shell, that is, CH2, CH, CH–CH3 and C–CH3, in addition to radicals with one unpaired electron surrounded by lone pair electrons, that is, OH, CH3O and CH3CH2O. These entities act indistinguishably from atoms as regards their stable binding with a high coordination number. The UBI-QEP model predicts chemisorption heats that in general can reach 120 kcal mol−1; 18 we used the following formula for its calculation

The third group represents molecules bonded to the surface with medium strength like monovalent radicals that have a unique unpaired electron: CH3, CH2CH3, CH2CH2OH, CH3CH2OH, HCO, CH3CO and CH2CHO. The reactive centre of these radicals is trigonal hybridized and the odd electron occupies the 2pz carbon atomic orbital. The UBI-QEP utilizes a simple interpolation based on the arithmetic average of the weak and the strong interactions represented by equations (7) and (8), which produces equation (10)
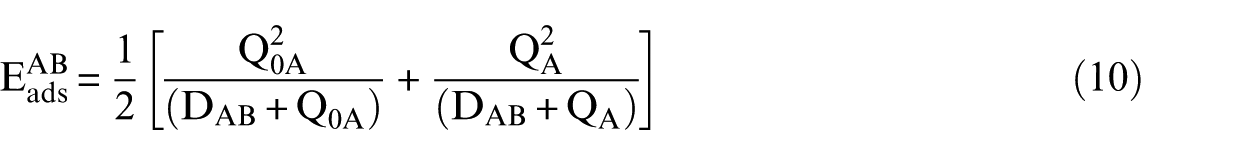
The contemporary concept of the UBI-QEP method does not determine the value of the AB bond index, while the system has to reach the transition state to be able to calculate the activation energy of an elementary step. It takes, however, an average value of the Lennard-Jones reaction barrier corresponding to surface adsorbed reactants. Then, we can write for the activation barrier of a quasi-diatomic molecule AB dissociated into A and B

where ΔH is the enthalpy of the surface reaction deduced from the thermodynamic cycle: desorption and gas-phase dissociation of A and then the chemisorption of A and B:
Outcomes and discussion
In this part, chemisorption energetics outcomes of ethanol dehydrogenation are presented. For each key specie, we highlight the dimensions of its optimized structure on the TM surfaces and adsorption energy of the most stable form. Thus, we inquire into the most probable reaction pathways as a means to discern trends of the reaction process on different TM surfaces.
Ethanol activation
Ethanol adsorption
Ethanol adsorption is the first step of its activation: it interacts with the top site of Cu(111), Ag(111), Ni(111), Pd(111) and Pt(111) surfaces through the oxygen lone pair of electrons, with the O–H bond parallel to the surface. This finding was observed by Xu et al. 20 using infrared reflection absorption spectroscopy (IRAS). In fact, the O–H stretching mode of the adsorbed ethanol species was absent in their spectra during the interaction of ethanol with Ni(111). They explained this by a plausible collateral orientation of the O–H bond relative to the surface. Also, methanol behaves in a similar manner when adsorbed on Pt(111). 21 Of the surfaces studied, the shortest ethanol surface distance was that of EtOH/Ni(111), where the functional H and O are situated at a distance of 2.357 and 2.144 Å from the surface, respectively.
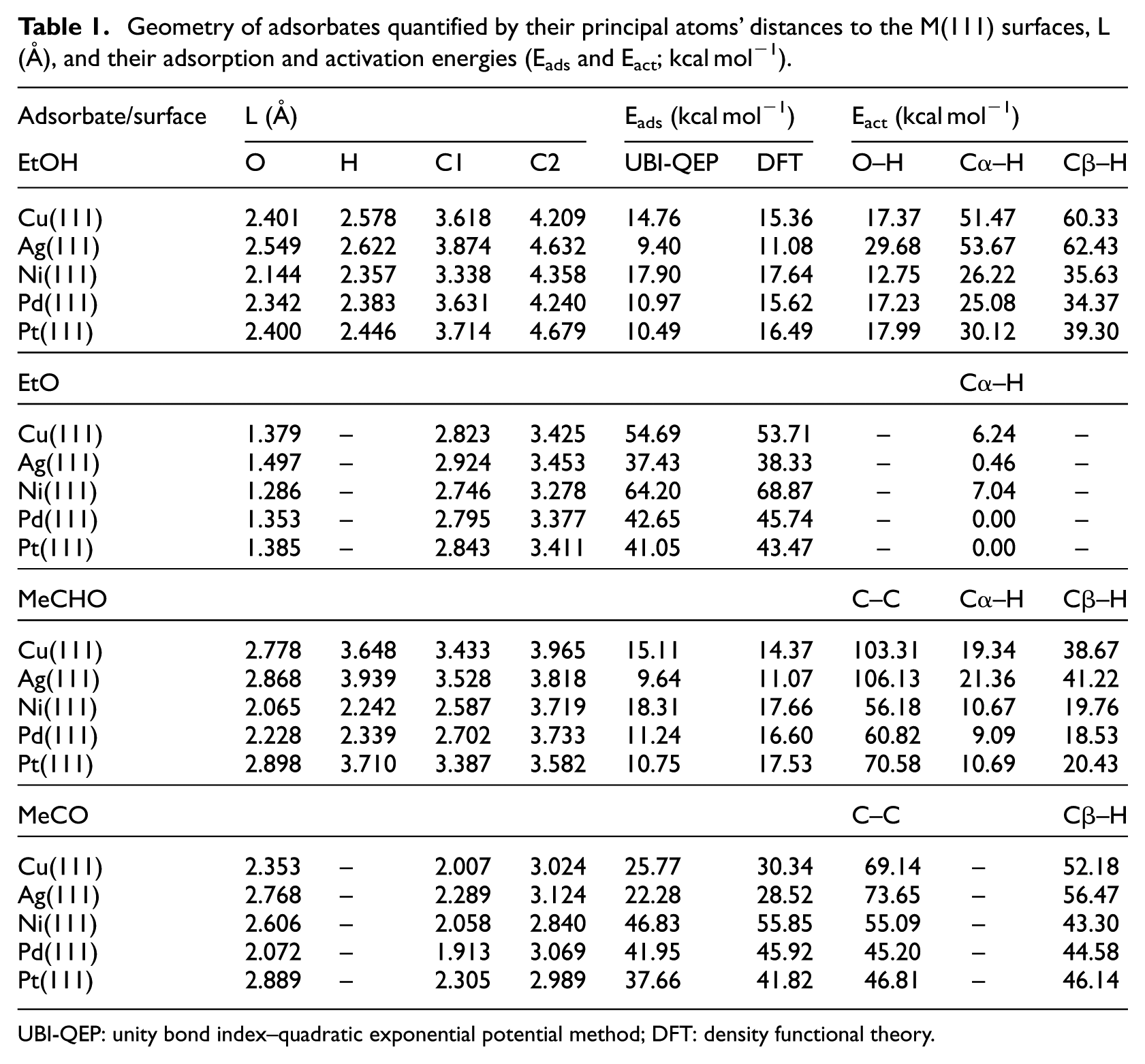
The computed adsorption energies of ethanol based on the UBI-QEP and DFT approaches are consistent with each other (Table 1). We note a very acceptable gap of 0.6, 1.68 and 0.26 kcal mol−1 for Cu, Ag and Ni, but a larger one for that of Pd and Pt with an average value of 5.3 kcal mol−1. Furthermore, a low adsorption energy that does not exceed 20 kcal mol−1 as the maximum value was noted for Ni(111): 17.90 kcal mol−1 indicates a weak interaction of ethanol with the TM surfaces. From an energetic point of view, our UBI-QEP results suggest that Co(0001) and Au(111) interact likewise with ethanol as with Ni(111) and Ag(111), respectively. By observing binding energy analysis with short adsorbate–surface distances, we can conclude that ethanol would undergo further transformation.
Geometry of adsorbates quantified by their principal atoms’ distances to the M(111) surfaces, L (Å), and their adsorption and activation energies (Eads and Eact; kcal mol−1).
UBI-QEP: unity bond index–quadratic exponential potential method; DFT: density functional theory.
We examined the electronic structure of two samples: EtOH/Cu(111) and EtOH/Pd(111) systems by plotting their local density of states (LDOS; Figure 1(a) and (b)). The peak of the d-band of the Cu atom that interacts with EtOH is barely altered after its adsorption, as its shape is conserved. In addition, the LDOS peaks associated with the p-orbital of the functional O atom are down shifted. However, the peaks corresponding to the adsorption state are still well defined compared to those of the gas-phase positions. 22 This gives another indication about the weak interaction of ethanol with the TM(111) surfaces.
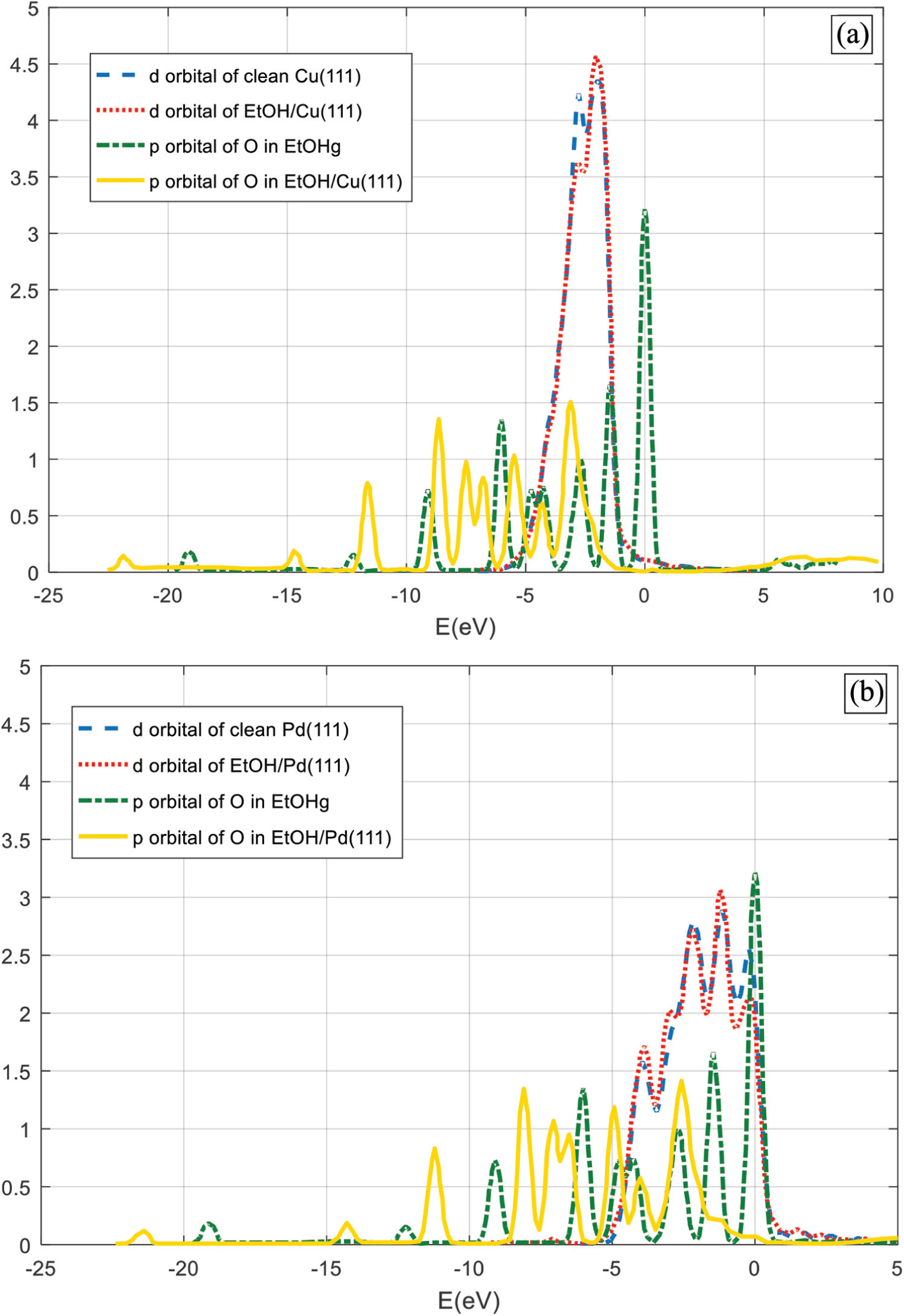
Local density of states (LDOS) of ethanol physisorbed on (a) Cu(111) and (b) Pd(111).
Ethanol transformation
We initiate this section by considering routes highlighted in experimental and DFT findings about ethanol activation on TM surfaces.20,23–25 All likely bond scission paths include Cα–H, Cβ–H and O–H in ethanol. We found after the computation of the activation energy for each pathway that there is a remarkable distinction among the different metals. Our UBI-QEP calculations predict that ethanol is most likely dehydrogenated via the O–H bond in a decreasing order from Ni, Co, Cu and Ag to Au as shown by Bell–Evans–Polanyi (BEP) and two-dimensional (2D) surface plots (Figure 2(a) and (b)). This is in total agreement with DFT and experimental observations.20,23–25 The temperature-dependent studies showed that ethanol is adsorbed as ethoxy on unsupported polycrystalline Cu, preserving Cα–H and Cβ–H except O–H. 6
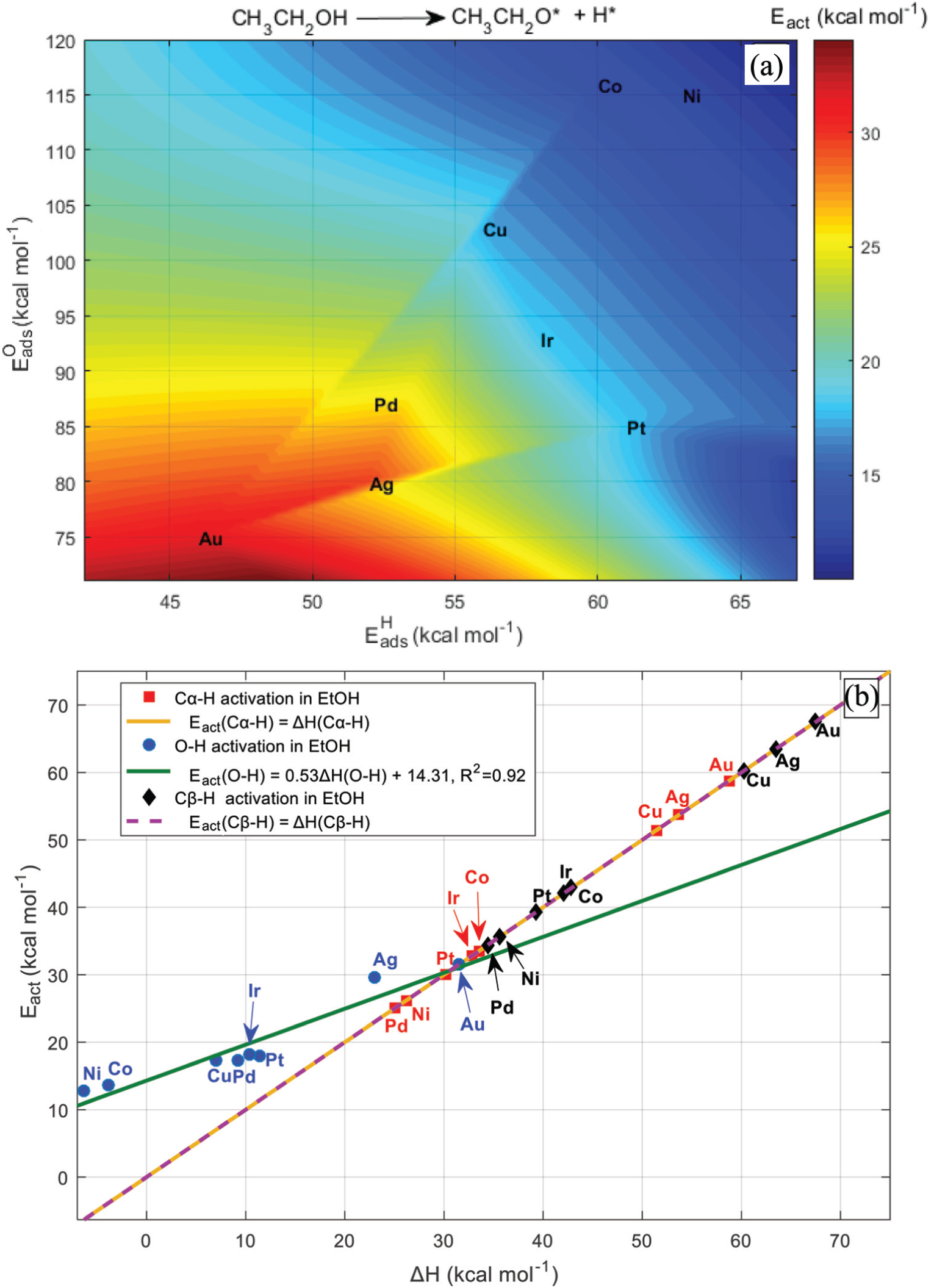
Activation energies of Cα–H, Cβ–H and O–H in EtOH: (a) O–H activation as a function of H and O binding energies,
Nevertheless, some DFT investigations suggest Cα–H breakage over Pd and Pt(111),26,27 while the adsorption–activation experiments indicated O–H bond cleavage. Even experimental results show discrepancies between the available studies concerning the scope of ethanol decomposition on Pt. Cong et al. 28 studied the decomposition of ethanol on Pt(331), the surface of which consists of (111) steps and (111) terraces. They observed decomposition via two separate pathways, both through C–C bond cleavage, and one of which, the low-temperature pathway entails ethanol C–C bond scission, followed then by dehydrogenation.
Ethoxy chemisorption
Geometry optimizations for all the studied ethoxy/(111) chemisorbed surfaces converge towards a stable structure for which its oxygen atom binds to the fcc three-coordination site. Other studies reported an equivalent high symmetry site, hcp, also to be stable.29,30 The C–O bond orientation coincides with a perpendicular vector to the (111) surface, this situation is confirmed by the observation of υas (CH2) intensity results from IRAS analysis. 20 The distance from its methyl group to the surface decreases from Ag, Cu, Pt and Pd to Ni.
Ethoxy species react strongly and similarly with Ni(111) and Co(0001) surfaces, and the chemisorption energy being almost 65 kcal mol−1for each. It is around 55 kcal mol−1 for Cu(111) and decreases to be about an average of 42 kcal mol−1 for Pd and Pt and then 34 kcal mol−1 for Au(111) (Table 1). This trend can be explained by the d-band theory, in which the d-band centre (εd) represents the reactivity index of TM surfaces. As stated in this model, an elevated εd, relative to the Fermi level, is associated with a decrease in filling of the anti-bonding states, leading to the stabilization of the metal-adsorbate system. Hence, there is an increased chemisorption energy. 31
The LDOS of the O atom in EtO and one of the M atoms of Cu(111) and Pd(111) to which it binds is plotted in Figure 3(a) and (b). The two peaks of the O p-orbital found in the gas phase have significantly shrunk on interaction with the Cu and Pd(111) surfaces.
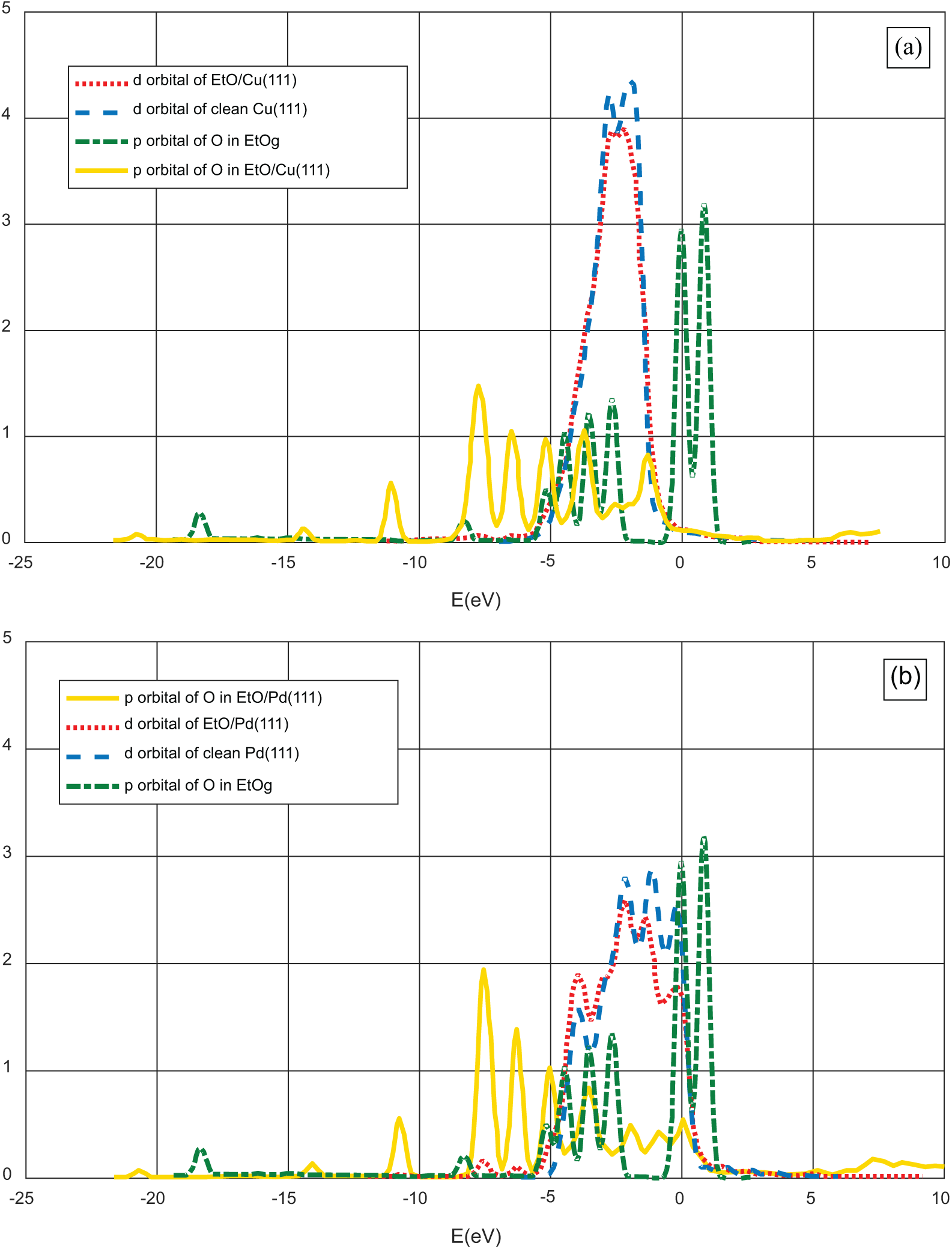
Local density of states (LDOS) of ethoxy, chemisorbed on (a) Cu(111) and (b) Pd(111).
The adsorbed ethoxy is effortlessly dehydrogenated to acetaldehyde over Pd, Pt, Ag, Au, and Ir, accompanied with a degree of exothermicity. Also, this step is almost athermic and it needs only an activation barrier of 6–9 kcal mol−1 over Cu, Ni and Co (Figure 4). Indeed, ethoxy is strongly restrained by the three 3d metals, while its chemisorption energy is relatively moderate with all the other surfaces. This behaviour affects the kinetics of ethoxide transformation that controls the selective dehydrogenation of ethanol to acetaldehyde, in addition to the interaction and the desorption of adsorbed acetaldehyde as well. In other words, a catalyst with an enhanced selectivity to acetaldehyde for this dehydrogenative process would have an impact on the rate of acetaldehyde formation in respect of its desorption rate.
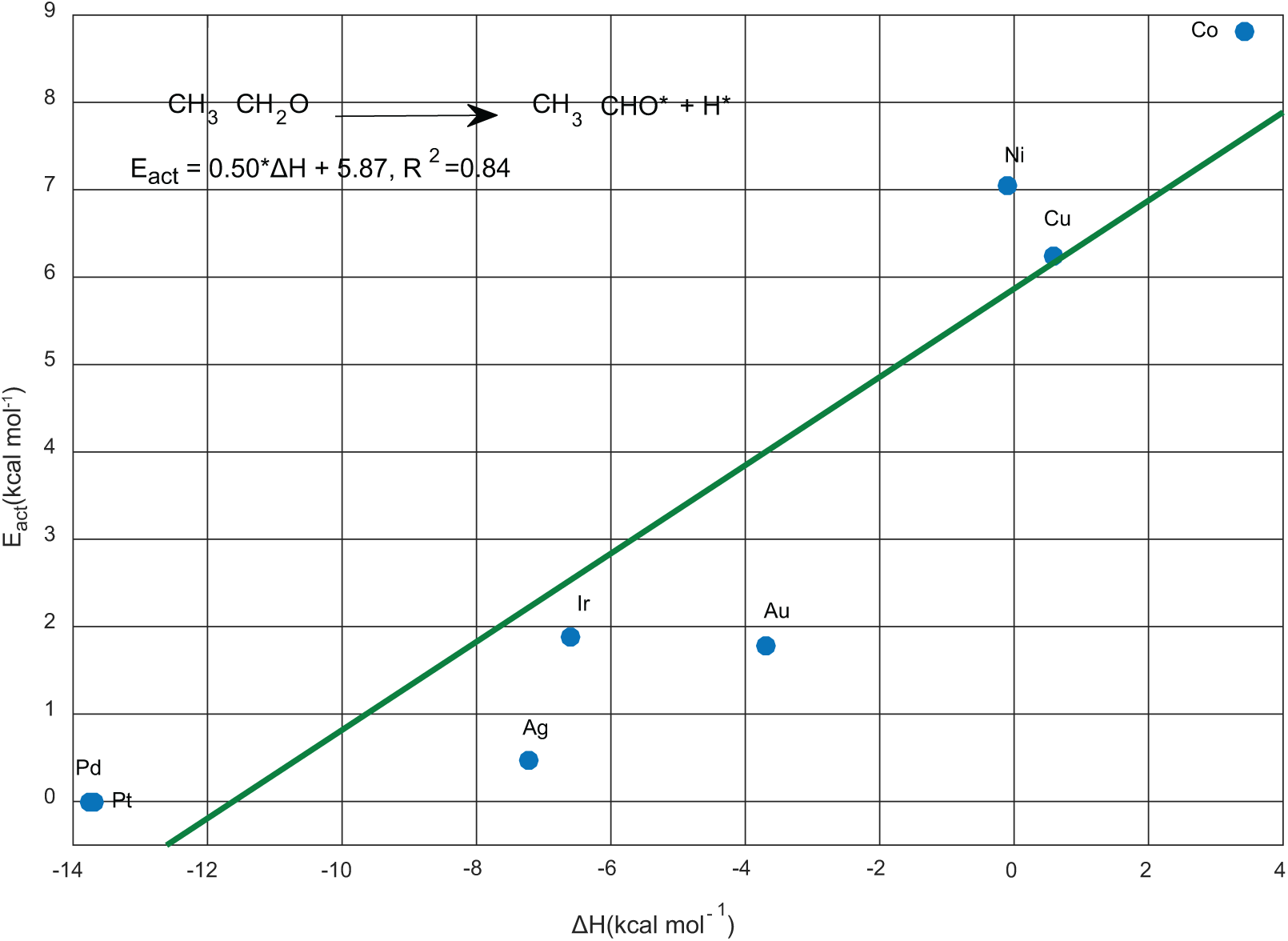
Bell–Evans–Polanyi (BEP) linear relationship for Cα–H activation in ethoxy.
Interaction of acetaldehyde with TM surfaces
Acetaldehyde adsorption
Because acetaldehyde is a key intermediate in ethanol dehydrogenation, its interaction with TM surfaces could affect the selectivity towards EA. Our calculations demonstrate that acetaldehyde adsorbs on Ag(111) and Cu(111) surfaces by interacting with its oxygen lone pair electrons in a η1 (O) configuration. However, for Pt(111), Pd(111) and Ni(111), the bonding is through a η2 (C, O) configuration where functional carbon and oxygen atoms coordinate with surface metal atoms over a bridge site. The calculated adsorption energies of acetaldehyde on Pt(111), Pd(111) and Ni(111) were 17.66, 16.60 and 17.53 kcal mol−1, respectively, higher than those of Ag and Cu (11.07 and 14.37 kcal mol−1). In fact, for a η2 coordination, electron transfer from d-orbital of the metal to anti-bonding orbitals of acetaldehyde C = O bond reinforces and stabilizes the metal-C bond. Nevertheless, electron contribution from the oxygen lone pair orbitals in the η1 coordination creates a weaker bond. This fact can be highlighted by analysing the LDOS as mentioned previously (Figure 5). For MeCHO/Cu(111) sample, the d-band of the interacted Cu is invariant, also the peaks of p-orbitals associated with the functional O atom are slightly shifted towards the left conserving its shape. Contrariwise, for MeCHO/Pd(111) sample, the second low energy peak of p-orbital of O is diminished with a small deformation of the d-band of the interacted Pd atom, giving a higher interacting energy. It is worth to note that the differences between the adsorption energies calculated by UBI-QEP and DFT of the MeCHO/Pd(111) and MeCHO/Pt(111) systems are due to the initial approximation for which we assumed that acetaldehyde interacts only with oxygen lone pair with Pd(111) and Pt(111) surfaces.
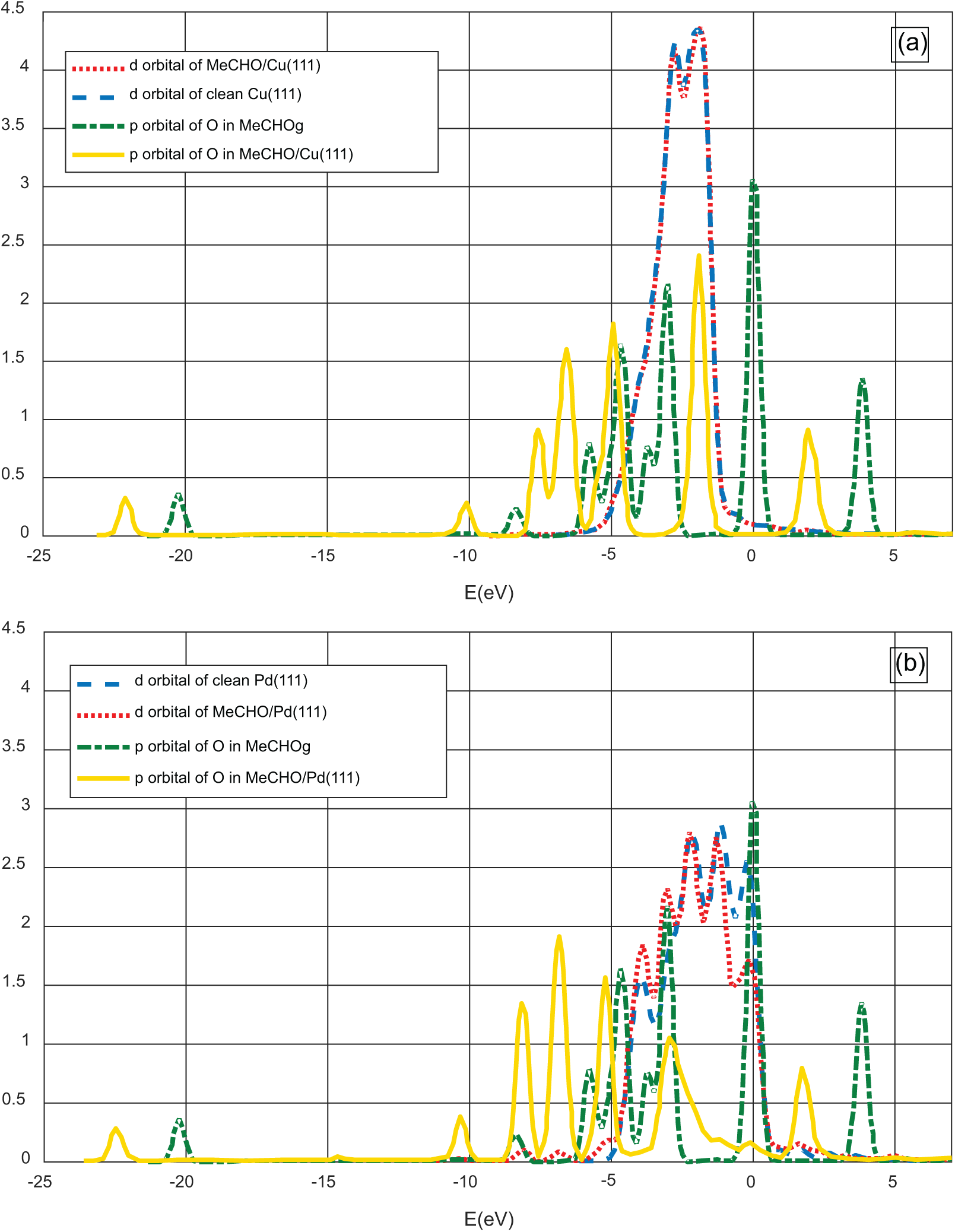
Local density of states (LDOS) of acetaldehyde interaction with (a) Cu(111) and (b) Pd(111).
Acetaldehyde transformation
For the case of acetaldehyde transformation, we distinguished bond-breaking pathways involving Cα–H, Cβ–H and C–C. We noted that all the studied surfaces show a tendency towards Cα–H scission leading to the co-adsorption of CH3CO and H. By comparing between the adsorbate/TM surfaces, we can subdivide them into two classes: the first includes Pd, Ni, Pt, Ir and Co, for which the activation energy varies between 9 and 13 kcal mol−1. This changes from 19 to 26 kcal mol−1 for the second class, associated with Cu, Ag and Au. In fact, this classification follows the adsorption conformation of CH3CHO on the TM surfaces. In contrast to the η1 (O) arrangement, in η2 (C, O) reinforcement of C = O, metal–C and metal–O interactions lower the stability of Cα–H and gives rise to a more labile H. The predicted mechanistic details of CH3CHO adsorption also affect the thermal characteristics of its chemical reaction with the (111) surfaces, as this process is exothermic for the Pd group surfaces, while it is relatively highly endothermic for the Cu group. Our UBI-QEP calculations for C–C and Cβ–H bond dissociations show that their energy barriers are larger than that of Cα–H, so such bond activation is unfavourable (Figure 6).
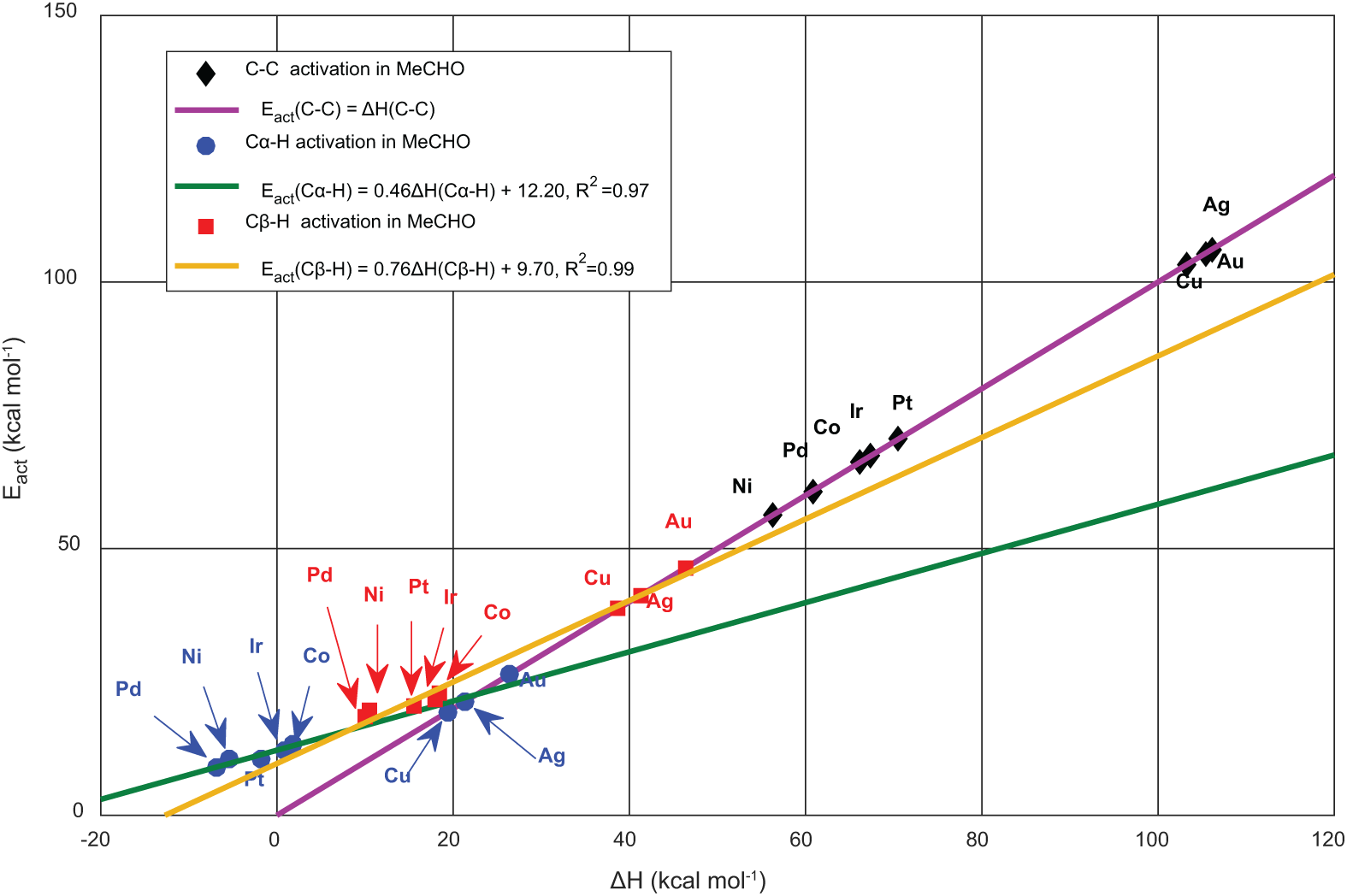
Bell–Evans–Polanyi (BEP) relations of Cα–H, Cβ–H and C–C activation in acetaldehyde.
Acetyl chemisorption
The top site is the most stable for acetyl chemisorption on (111) surfaces; thus, its Cα atom is strongly bonded to a surface metallic atom and its C = O axis is nearly aligned to the surface. The C-Pd length, 1.91 Å, was found to be the shortest distance of the investigated acetyl/(111) surfaces. The chemisorption energies of CH3CO on TM(111) surfaces decrease from Ni, Co, Pd, Ir to Pt and for the second set from Cu, Ag to Au (Table 1).
Regarding the decomposition of acetyl, two bond scissions within the bounds of possibility are considered: C–C and Cβ–H bonds. Our UBI-QEP results show that Cu, Ag and Au(111) surfaces stabilize the CH3CO species noting that energy barriers for C–C and Cβ–H breakage vary from 69 to 74 kcal mol−1 and from 52 to 62 kcal mol−1, respectively (Figure 7). The remaining surfaces Ni, Co, Pd, Pt and Ir tend to activate the two bonds. The calculated activation energies of C–C and Cβ–H on Pd and Pt surfaces are equivalent. The experimental examination of a kinetic isotope effect indicated that acetyl transformation on Pd(111) begins by carbon–hydrogen bond splitting, followed by a swift carbon–carbon bond breaking to yield CO and adsorbed monohydrocarbon species. 32 DFT calculations nominate Cβ–H to be more likely to occur. 26 Iwasa et al. remarked that over metallic Pd, the decomposition of ethanol into carbon monoxide, methane and hydrogen took place to a significant degree. They demonstrated that Pd alloys stabilize acetaldehyde, which facilitates its transformation into EA by the interaction with ethanol. However, over metallic Pd, acetaldehyde undergoes decarbonylation to CO and CH4. 33
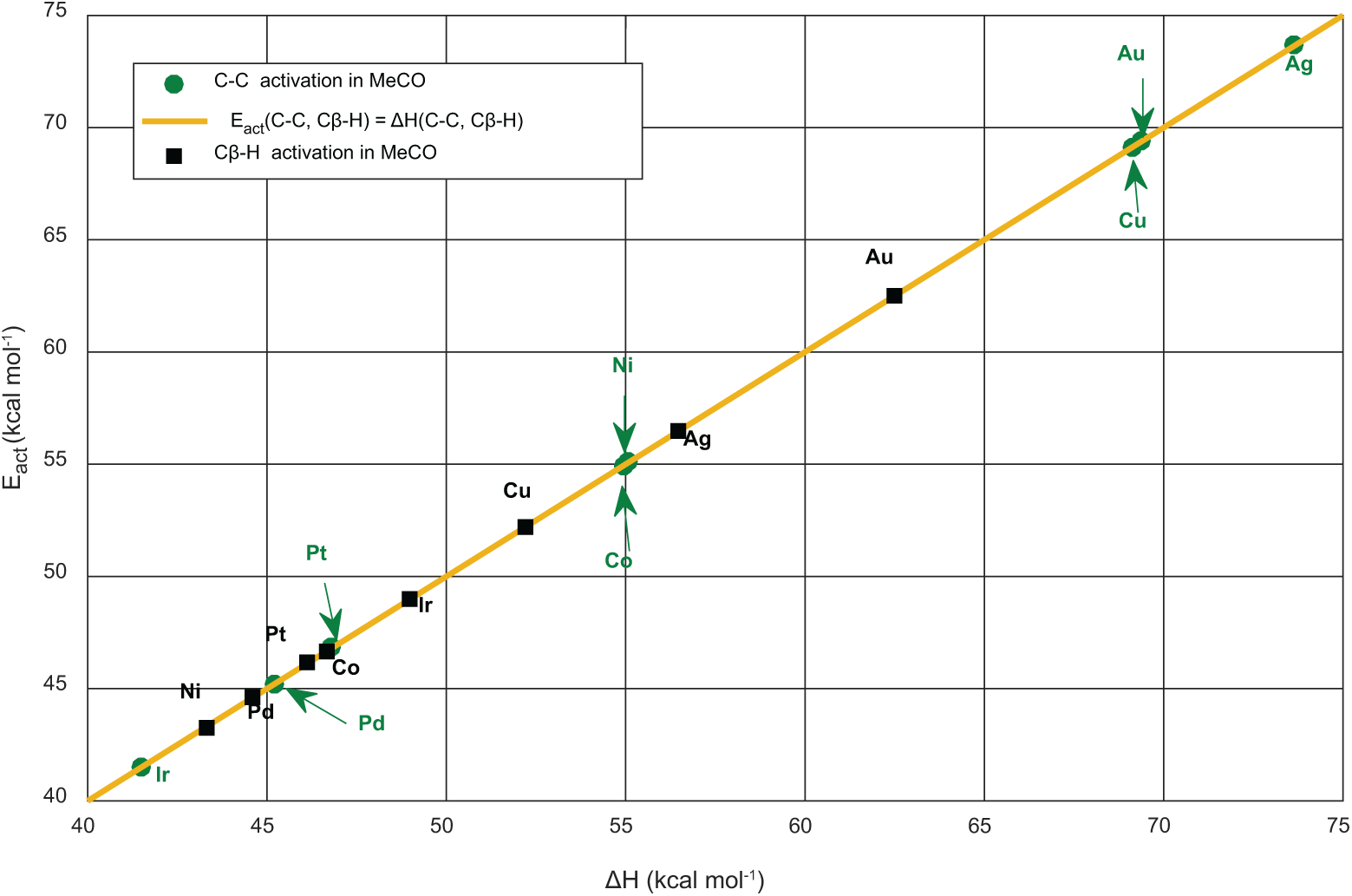
Bell–Evans–Polanyi (BEP) relations of Cβ–H and C–C activation in acetyl.
EA formation
EA adsorption
The optimized structure of adsorbed EA involves bonding through the carbonyl oxygen to a lone metallic surface atom. The distance from the O of C = O to the surface was the shortest for EA/Ni(111), that is, about 2.263 Å, and the second shortest distance was 2.379 Å for EA/Pd(111). Besides, our findings point out that the CCCC-axis of EA is approximately parallel to the (111) surfaces, as concluded theoretically by Li et al. 34 for EA/Cu(111). Noting that Zahidi et al. 35 studied experimentally the decomposition of methyl acetate (MA) on Ni(111), they deduced that MA adsorption results through the interaction of the carbonyl lone pair with the metal.
However, the direction of the molecular plane tends to be approximately perpendicular to the surface. Energetically, DFT and UBI-QEP results show that EA interacts weakly with all (111) surfaces (Table 2). The calculated value of adsorption energies varies between 7 and 13 kcal mol−1. These weak energies reflect the fact that the functional atoms of EA interact weakly with all the (111) surfaces.
EA principal atoms’ distances to the M(111) surfaces, L (Å); its adsorption and formation energies (Eads, Eact) (kcal mol−1). C= and O= atoms are C and O in EA’s C=O. C– and O– are C and O in −C–O of EA.
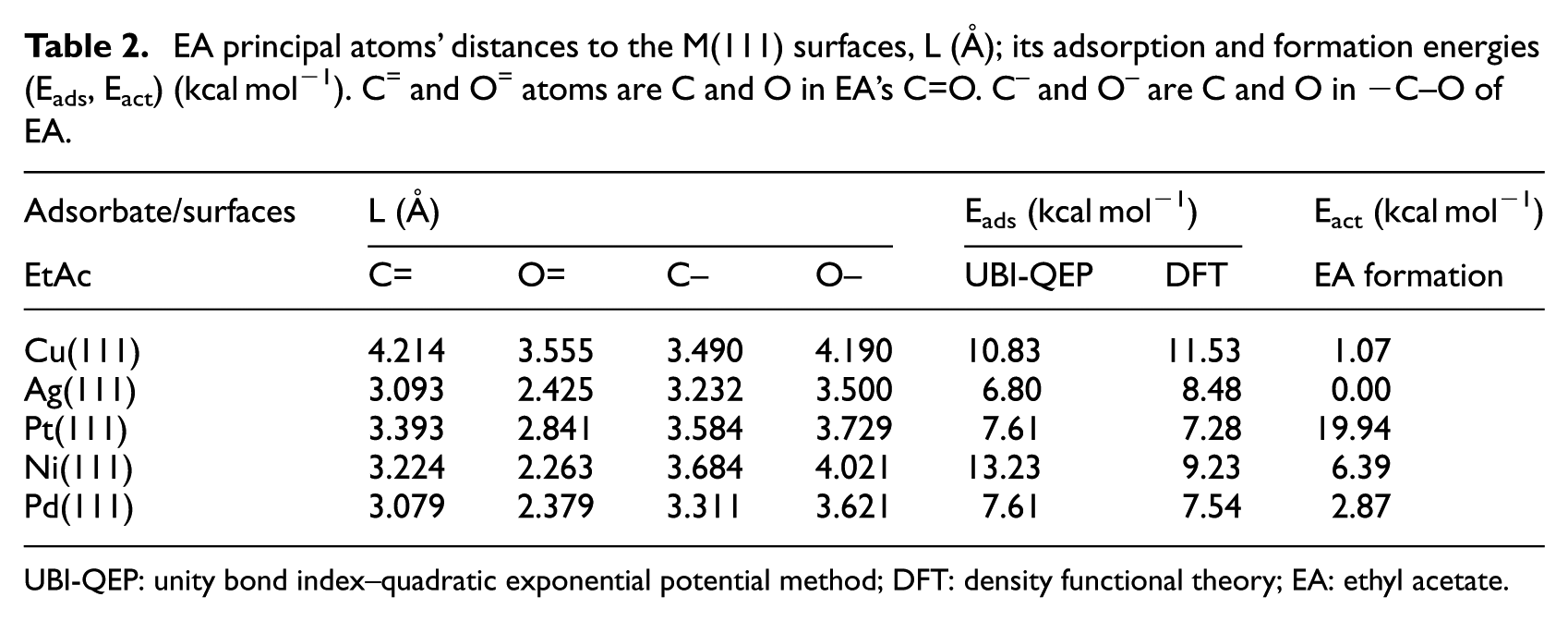
UBI-QEP: unity bond index–quadratic exponential potential method; DFT: density functional theory; EA: ethyl acetate.
EA formation barriers
The formation of EA starting from the adsorbed ethoxy and acetyl species represents the last step in the ethanol dehydrogenative process to EA. We can remark that all the studied surfaces activate the combination between EtO and MeCO with moderate energy barriers, as shown in Figure 8. The catalytic behaviour of the studied surfaces can be related to retention of the key intermediates, quantified by their chemisorption energies (Figure 8(a)). Hence, we subdivide the TM(111) surfaces into three groups. The first one, which has relatively high activation barriers of 18 and 20 kcal mol−1, associated with an exothermic effect of ‒9 and ‒13 kcal mol−1 for Co and Ni, respectively. The second group involved Cu, It, Pd and Pt. The interaction of EtO and MeCO with these surfaces is quite moderate, as shown from their chemisorption energies. Having a balance between retention and desorption gives a compromise, allowing intermediates to diffuse on the surface. The third group is composed of Au and Ag, on which the association reaction of ethoxy and acetyl is spontaneous. However, the thermal effect is endothermic. The binding energies of EtO and MeCO to Au and Ag(111) are low; this increases the probability of desorbing the reactants before their reaction. It is notable that Pt and especially Cu possess optimal binding energies of the reactants, overcoming the shortcomings of the other surfaces studied.
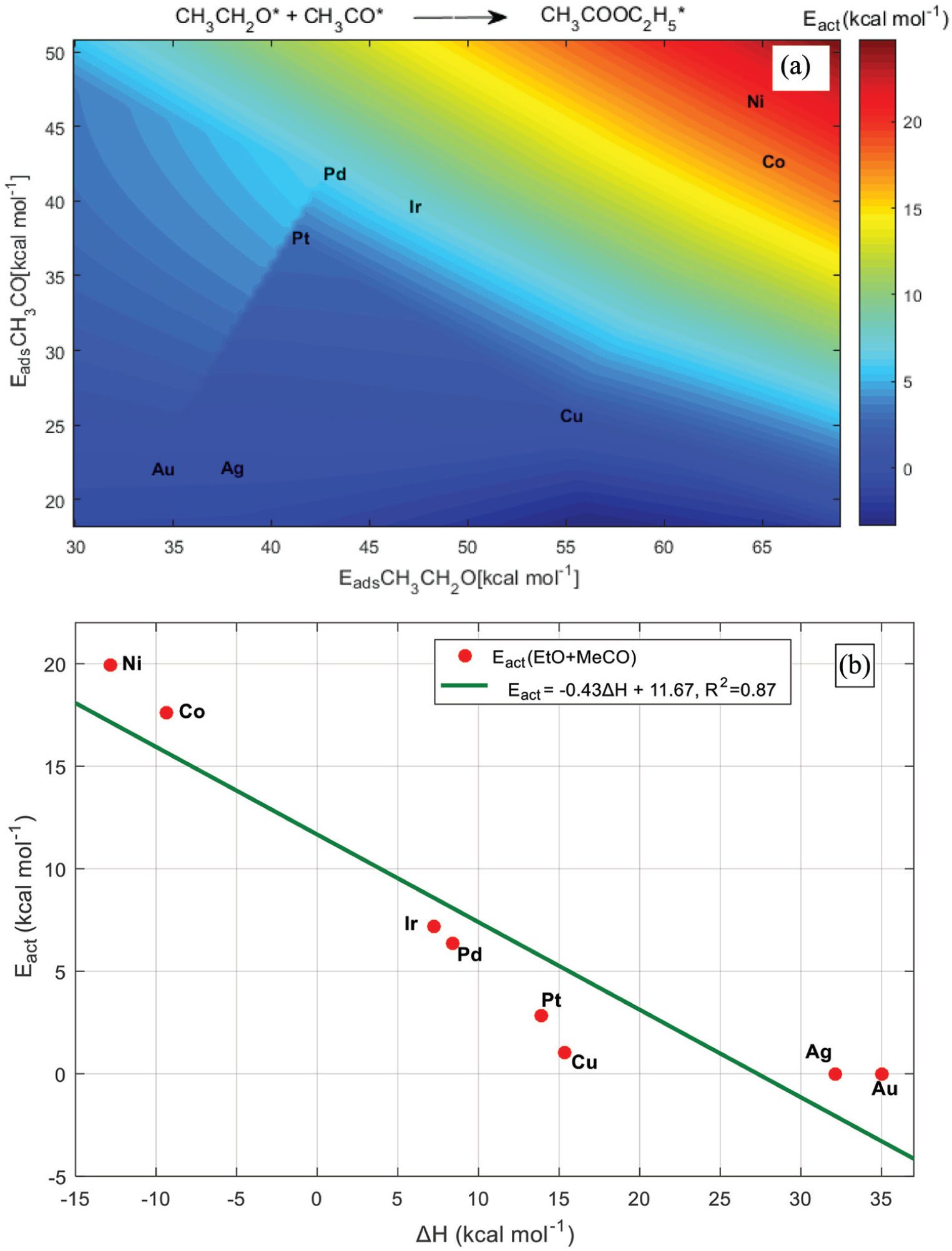
Activation energy of the combination between ethoxy and acetyl: (a) activation energy as a function of EtO and MeCO binding energies,
Conclusion
By combining DFT and UBI-QEP calculations, we have studied the interaction energetics of the dominant intermediates involved in the elementary reaction steps of ethanol dehydrogenative route to EA on Cu, Ag, Ni, Pd, Pt, Co, Au and Ir(111) surfaces. For all the studied TMs, the first bond scission in ethanol is through O–H, more easily on 3d metal surfaces. Acetaldehyde binds to Cu, Au and Ag through an η1 configuration, but Ni, Pd and Pt surfaces induce pi back bonding leading to η2. The η2 binding mode attracts acetaldehyde species and weakens their Cα–H bond, facilitating acetyl generation. We deduce that alloying Cu with Ni, Co, Pd, Pt or Ir may enhance the dehydrogenative route to EA. Our results show that the TMs studied can be divided into three groups regarding their catalytic behaviour towards the ethoxy and acetyl reactive species. The first group includes Ni and Co, for which the high adsorption energies of ethoxy and acetyl lead to their high retention. Second, Cu, and to a much lesser extent, Ag and Au, stabilize the reactive intermediates. This may explain the high catalytic activity of Cu-based catalysts for ethanol dehydrogenation to acetaldehyde and EA. The last group, Pd and Pt surfaces, tends to dehydrogenate acetyl intermediates, while Ir stimulates their decarbonylation.
Supplemental Material
PRK1900772_ESI_supplementary_text – Supplemental material for Interaction of intermediates with transition metal surfaces in the dehydrogenation of ethanol to ethyl acetate: A theoretical investigation
Supplemental material, PRK1900772_ESI_supplementary_text for Interaction of intermediates with transition metal surfaces in the dehydrogenation of ethanol to ethyl acetate: A theoretical investigation by Adel Boualouache, Ali Boucenna and Ghazi Otmanine in Progress in Reaction Kinetics and Mechanism
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship and/or publication of this article.
Supplemental material
Supplemental material for this article is available online.
Author biographies
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.