
Abstract
By performing density functional theory calculations, the adsorption configurations of formic acid and possible reaction pathway for HCOOH oxidation on PtPd(111) surface are located. Results show that CO2 is preferentially formed as the main product of the catalytic oxidation of formic acid. The formation of CO on the pure Pd surface could not possibly occur during formic acid decomposition on the PtPd(111) surface owing to the high reaction barrier. Therefore, no poisoning of catalyst would occur on the PtPd(111) surface. Our results indicate that the significantly increased catalytic activity of bimetallic PtPd catalyst towards HCOOH oxidation should be attributed to the reduction in poisoning by CO.
Introduction
The direct formic acid fuel cell (DFAFC) appears to be one of the promising clean energy sources due to its high energy conversion efficiency and low environmental pollution. 1 Its direct oxidation releases two electrons per molecule 2
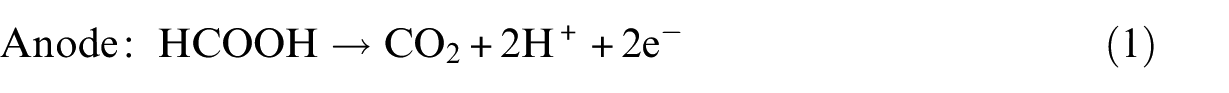
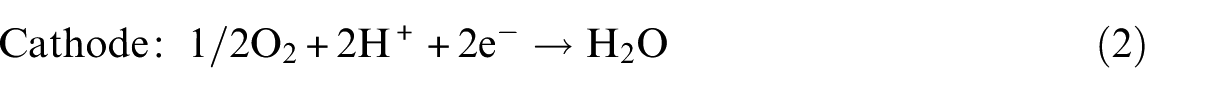
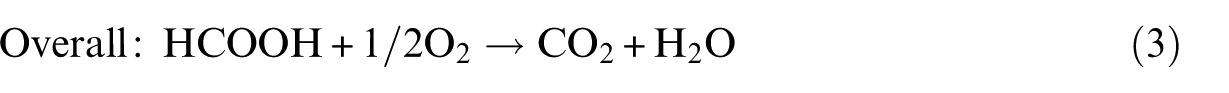
It is generally accepted that the mechanism of the formic acid oxidation to CO2 follows dual pathways,3,4 that is, the dehydration pathway via adsorbed CO (HCOOH → CO + H2O → CO2 + 2 H+ + 2e−) or the dehydrogenation pathway without the participation of CO (HCOOH → CO2 + 2 H+ + 2e−). In DFAFC, the sluggishness of formic acid oxidation on the anode catalyst is one of the primary bottlenecks. In general, Pd-based catalysts are commonly used in DFAFC. 5 However, the catalytic performance of pure Pd catalyst would be significantly diminished owing to the poisoning effect of CO generated from the HCOOH dehydration pathway. 3 Therefore, a highly selective and active catalyst towards the HCOOH dehydrogenation pathway 4 with formation of H2 and CO2 is desirable.
Recent studies showed that modification of the Pd surface with a secondary metal, such as Ni,6,7 Au,8,9 Co 10 and Ag,11,12 is considered to be an efficient way to hinder the dehydration pathway, 13 by taking advantage of the so-called electronic ligand effect 14–16 and the geometrical ensemble effect.14,15,17,18 Among these Pd-based bimetallic catalysts, Pt-Pd nanostructures have attracted special attention due to their higher tolerance to CO poisoning and better catalytic activity compared with pure Pd catalysts. 19 Recently, the Pt-modified Pd catalyst has been prepared as the electrocatalyst for the HCOOH oxidation, such as Pt-decorated Pd/C nanoparticles, 20 Pt sub-monolayers–decorated Pd black, 21 Pt-decorated Pd nanochain networks 22 and carbon-supported Pd-Pt alloy nanoparticles. 23
Although much is known about the preparations of different morphological Pt-modified Pd catalysts, the underlying mechanism of their improved CO-poisoning tolerance still remains unclear at the molecular level. Herein, this study presents a theoretical study of formic acid oxidation on the bimetallic PtPd(111) surface. The (111) surface is considered as the most stable crystal plane of the fcc lattice and therefore is expected to contribute the most significant areas for the surface of bulk metals, nanoparticles and alloys and thus has been chosen for the present work. By performing the ultrasoft pseudopotential 24 density functional theory (DFT) calculations, we have shown the mechanistic details of the elementary steps of HCOOH oxidation on a PtPd(111) surface, from which we hoped to elucidate the reason why Pt-Pd bimetallic catalysts possess enhanced catalytic activity and improve the CO-poisoning tolerance.
Computational details
The PtPd(111) surface was obtained based on the Pd(111) surface, where three Pd atoms in the top layer were substituted by three Pt atoms. In such a PtPd(111) surface model, the Pt atoms in the top layer were isolated by the Pd modifier and fixed to Pd(111) to follow the idea of maximizing the use of the efficiency of Pt materials. Our calculations were performed by using a triple-layer p(3 × 3) unit cell, where the atoms in the top layer were allowed to relax, whereas those in the two bottom layers were fixed. The vacuum region between slabs was 10 Å, which was sufficiently large to guarantee that the interactions between neighbouring cells in a direct normal to the surface were negligible.
All DFT calculations were performed using the CASTEP code.25,26 The exchange-correlation effects were described with the spin polarization generalized gradient approximation (GGA) using the Perdew–Burke–Ernzerhof (PBE) functional, 27 which has often been used to investigate small molecule oxidation on noble metal surfaces and the calculated results are reliable. 28 Our calculated results show that the lattice constant is 3.92 Å for the bulk Pd, which is in good agreement with the experimental value of 3.89 Å. 29 A mesh of 2 × 2 × 1 Monkhorst–Pack special k-point grid was used for the integrations of the Brillouin zone. The electron wave functions were expanded by a plane-wave basis set with a cut-off energy of 400 eV. The criteria for maximum force convergence and energy were set to 0.05 eV Å−1 and 2.0 × 10−5 eV atom−1. The transition states (TSs) were determined with the linear and quadratic synchronous transit (LST/QST) method. 25
The adsorption energies for HCOOH were defined as Ead = Esurf + Eadsorbate − Etotal, where Esurf, Eadsorbate and Etotel refer to the energies of the clean surface, free formic acid molecule and the adsorbed system, respectively. The activation barrier energy (ΔEa) of each elementary step was computed by ΔEa = ETS − ER, where ETS and ER are the energies of the transition state and reactant, respectively.
Results and discussion
Formic acid adsorption on PtPd(111) surface
The adsorption of formic acid on heterogeneous catalysts is the first and governing step in the process of oxidation. It should be noted that formic acid has two isomers: transoid-HCOOH and cisoid-HCOOH. Initially, our results showed that the transoid-HCOOH is more stable than the cisoid one in the gas phase by14.5 kJ mol−1, which is consistent with the experimental finding of 16.4 kJ mol−1.
30
We then examined the adsorption of the two isomers of HCOOH on the PtPd(111) surface, including top, bridge, fcc and hcp sites. Figure 1 shows four adsorption configurations, labelled as
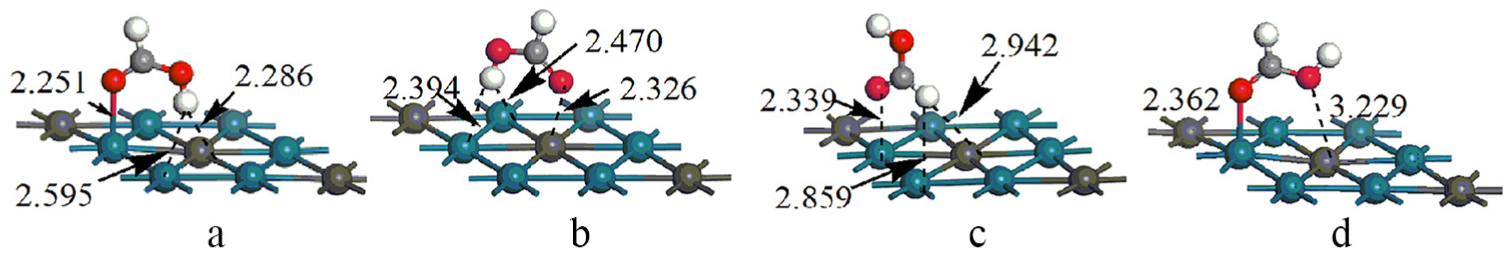
Optimized adsorption configurations of HCOOH on PtPd(111) surface with crucial structural parameters (in Å).
From our calculations, the above three adsorption structures of HCOOH on the PtPd(111) surface are similar to those on the Pd(111) surface shown in our previous study. 3 However, the adsorption energies of HCOOH seem to decrease compared to those of their counterparts (42.5, 37.7 and 19.3 kJ mol−1 on the PtPd(111) surface and 59.8, 36.7 and 40.5 kJ mol−1 on the Pd(111) surface). Generally, the d-band centre shift reported by Norskov and colleagues32,33 can be used as the simplest descriptor to measure the variation of the interaction of surface atoms with adsorbates: the d-band centre moving up towards the Fermi level can strengthen the binding energies of adsorbates on catalyst surfaces. In this study, the d-band centre for the Pt/Pd(111) surface was calculated to up-shift to −0.19 eV from −1.86 eV for the Pd(111) surface. However, the trend of the calculated adsorption energies shows deviations from the d-band model. Such unexpected results are due to the modification of the local d-band by the presence of the adsorbate, which is similar to that found in previous researches.34,35
Formic acid oxidation on PtPd(111) surface
The oxidation of formic acid to CO2 would produce hydrogen atoms and electrons. The atomic charge population of H+ and Au surface was first calculated to understand the electronic structure of adsorbed H+ on Au surface. When H+ is adsorbed on an Au surface, the electron transfers from Au surface to H+. From our calculation, the atomic charge population of adsorbed H+ is close to that of H. Moreover, the metal acts as an electron sink and the transferred electron from Au to H is used as an electric power during the DFAFC operation condition. As a result, we assume the neutral model for the slab. We then present the possible pathways for formic acid oxidation. The optimized structures of the reaction intermediates and products located on the path are summarized in Figure 2, while the calculated energy profiles are shown in Figure 3, where the sum of the energies of the isolated reactants (HCOOH and the clean PtPd(111) surface) is taken as zero energy.
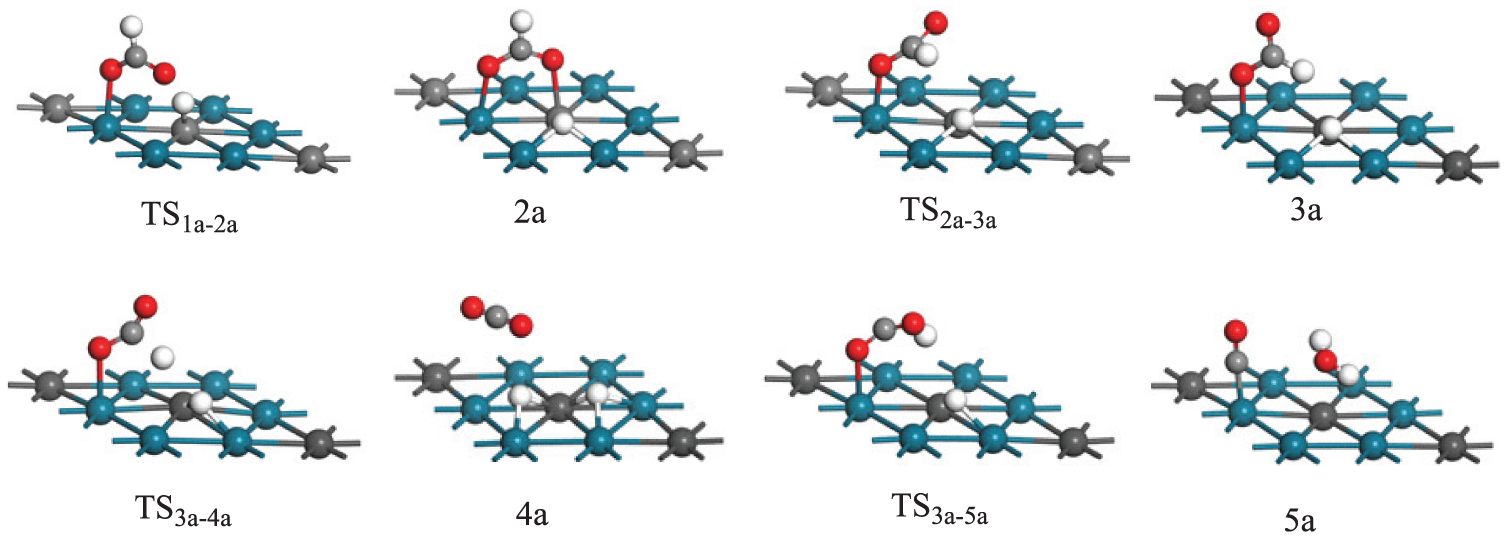
Optimized structures of intermediates and transition states. Bond lengths are in angstroms.
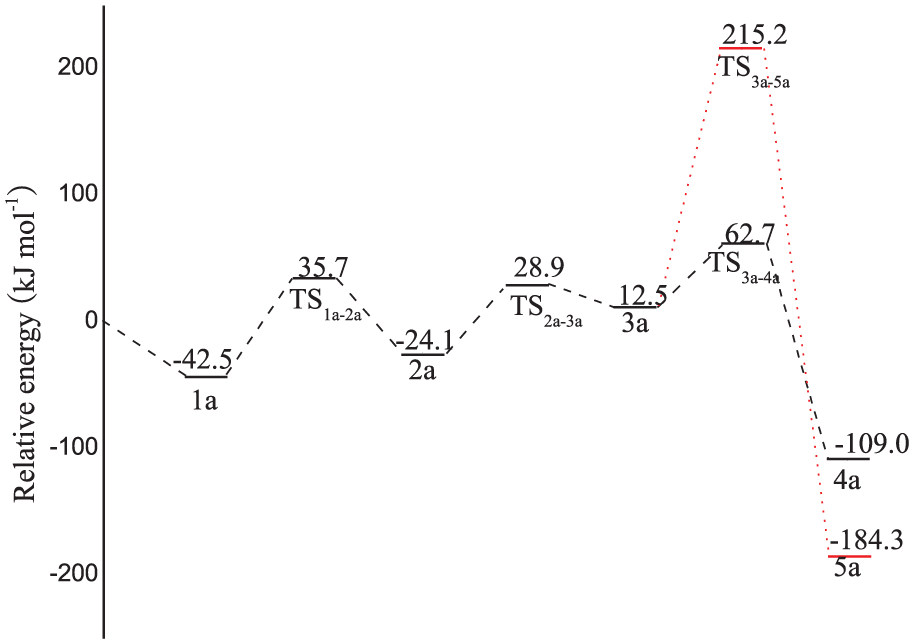
Potential energy profiles for HCOOH oxidation.
The stable configuration
To better understand the increased catalytic activity of bimetal PtPd catalysts, the activation energy barriers are compared with our previous results 3 for the monometallic Pd(111) surface. On one hand, for the dehydrogenation process of HCOOH to CO2 on Pd(111), the transformation of HCOO from the bidentate structure to the monodentate structure is the rate-determining step, with a barrier of 83.9 kJ mol−1. However, the barrier of this step on the PtPd(111) surface is 53.0 kJ mol−1, which is not notably different from that on the Pd(111) surface, indicating that the PtPd(111) surface can perform similar activity for the catalytic dehydrogenation of HCOOH to CO2. On the other hand, we find that the barrier in the rate-determining step is 202.7 kJ mol−1 for the decomposition of HCOOH to CO on PtPd(111), whereas the corresponding result is 55.0 kJ mol−1 on Pd(111), implying that the CO pathway on the PtPd(111) surface is more difficult than that on a pure Pd(111) surface, which is in good agreement with the observed CO antipoisoning ability of PtPd bimetal catalysts for HCOOH oxidation. Given the present results, we propose that the enhanced catalytic activity of bimetal PtPd catalysts towards HCOOH oxidation should be attributed to their suppression to the dehydration pathway.
It is important to note that the reduced CO-poisoning effect of PtPd bimetallic is influenced not only by the relative rate of CO formation but also by the binding ability of CO on the surface. The stronger the binding of CO, the easier the poisoning of the catalyst. In order to understand the enhanced CO tolerant of PtPd catalysts, we calculated the adsorption energies of CO on PtPd(111) and Pd(111) surfaces, respectively. Our results showed that the adsorption energy of CO on the PtPd(111) surface is noticeably smaller than that on the pure Pd(111) surface (37.6 vs 377.3 kJ mol−1), implying that CO desorption from PtPd(111) is easier than that from Pd(111), also supporting the enhanced activity of PtPd bimetallic catalysts towards formic acid oxidation.
Conclusion
Formic acid oxidation on PtPd(111) surface has been investigated by DFT to provide a clue for the increased catalytic activity of PtPd bimetal catalysts. Comparing the activation energy barriers with Pd(111), our calculated results illustrate that PtPd(111) can perform as well as pure Pd for the catalytic dehydrogenation of HCOOH to CO2. On the contrary, HCOOH oxidation to CO on PtPd(111) becomes more difficult than that on the pure Pd(111) surface. Thus, the enhanced catalytic activity of bimetallic PtPd(111) surface towards HCOOH oxidation is owing to its suppression of the dehydration pathway.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received financial support for the research, authorship, and/or publication of this article: This research was financially supported by A Project of Shandong Province Higher Educational Science and Technology Programme (J18KA100) and Science and Technology Development Plan of Zibo (No. 2016kj010034).