
Abstract
X2Si=Sn: (X = H, Me, F, Cl, Br, Ph, Ar, etc.) are a new chemical species. The cycloaddition reactions of X2Si=Sn: are a new field of stannylene chemistry. The mechanism of the cycloaddition reaction between singlet state Me2Si=Sn: and ethene has been investigated for the first time here using second-order Møller-Plesset perturbation theory together with the 6-311++G** basis set for C, H and Si atoms and the LanL2dz basis set for Sn atoms. From the potential energy profile, it could be predicted that the reaction has one dominant reaction channel. The reaction process presented is that the 5p unoccupied orbital of Sn in Me2Si=Sn: and the π orbital of ethene form a π → p donor–acceptor bond resulting in the formation of an intermediate. The instability of this intermediate makes it isomerize to a four-membered Si-heterocyclic ring stannylene. Because the 5p unoccupied orbital of the Sn atom in the four-membered Si-heterocyclic ring stannylene and the π orbital of ethene form a π → p donor–acceptor bond, the four-membered Si-heterocyclic ring stannylene further combines with ethene to form another intermediate. Because the Sn atom in this intermediate assumes sp3 hybridization after the transition state, the intermediate isomerizes to a Si-heterocyclic spiro-Sn-heterocyclic ring compound. This result indicates the modes of cycloaddition reactions between X2Si=Sn: and symmetric π-bonded compounds, i.e. this study opens up a new field for stannylene chemistry.
Keywords
Introduction
The unsaturated olefins of the main group IV elements (C, Si, and Ge) are all reactive intermediates.1–6 Their cycloaddition reactions have been studied,7–11 and with the progress of these studies, study of cycloaddition reactions of unsaturated stannylene should also enter the agenda. However, there has been no published report about cycloaddition reactions of unsaturated stannylene until now. The unsaturated stannous species (e.g. X2Si=Sn: (X = H, Me, F, Cl, Br, Ph, Ar, etc.)) are a new chemical species. This opens up the possibility of research in a new field of stannylene chemistry, in particular the mechanisms of their cycloaddition reactions. To explore the rules of the cycloaddition reaction between X2Si=Sn: and symmetric π-bonded compounds, Me2Si=Sn: and ethene were selected as model molecules in this article, and the cycloaddition reaction mechanism was investigated and analyzed theoretically. The result indicates the modes of the cycloaddition reactions between X2Si=Sn: and symmetric π-bonded compounds, which are significant for the synthesis of small rings with Si and Sn, and Si-heterocyclic spiro-Sn-heterocyclic ring compounds. This study thus extends the research area and enriches the profile of stannylene chemistry.
Calculation method
We used second-order perturbation theory (MP2) 12 and the Gaussian 09 package to optimize the structure of Me2Si=Sn, its cycloaddition reaction with ethene and its transition states at the MP2 level of theory together with the 6-311++G** basis set for C, H and Si atoms and the LanL2dz basis set for Sn atoms. In order to confirm further the correctness of the relevant species and get the thermodynamic functions for the species, vibrational analysis was included. Finally, the intrinsic reaction coordinate (IRC)13,14 was also calculated for all the transition states to determine the reaction paths and directions.
Results and discussions
The energies of the singlet and triplet states of Me2Si=Sn: (R1) calculated at the MP2 level of theory are −371.84065 and −371.83888 au, respectively. So, the ground state of Me2Si=Sn: is a singlet state. On this issue, Bundhun et al. 15 also made relevant reports. Theoretical research shows that the cycloaddition reaction between singlet state Me2Si=Sn: and ethene has the following two possible pathways.
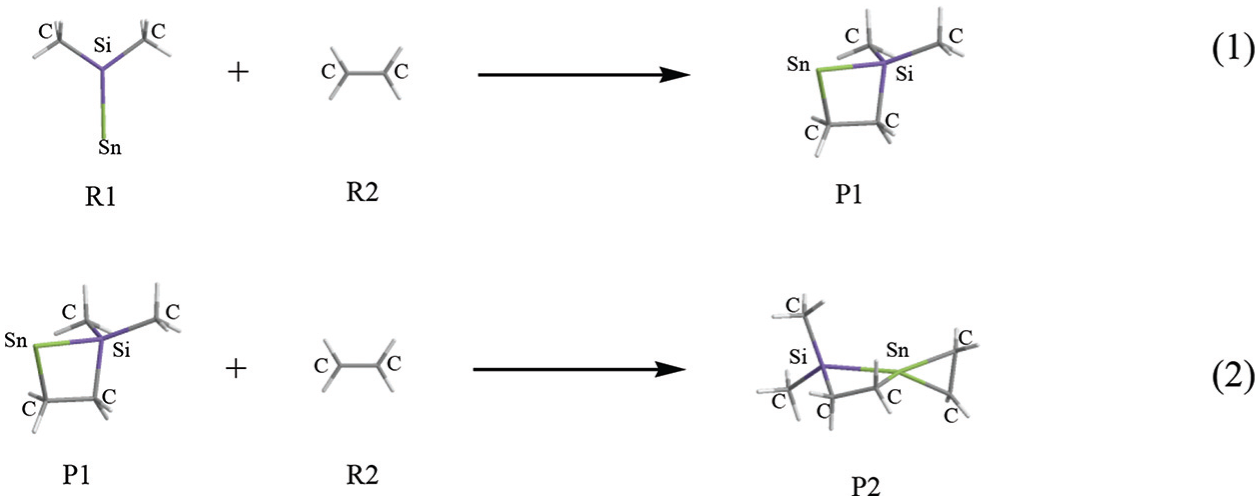
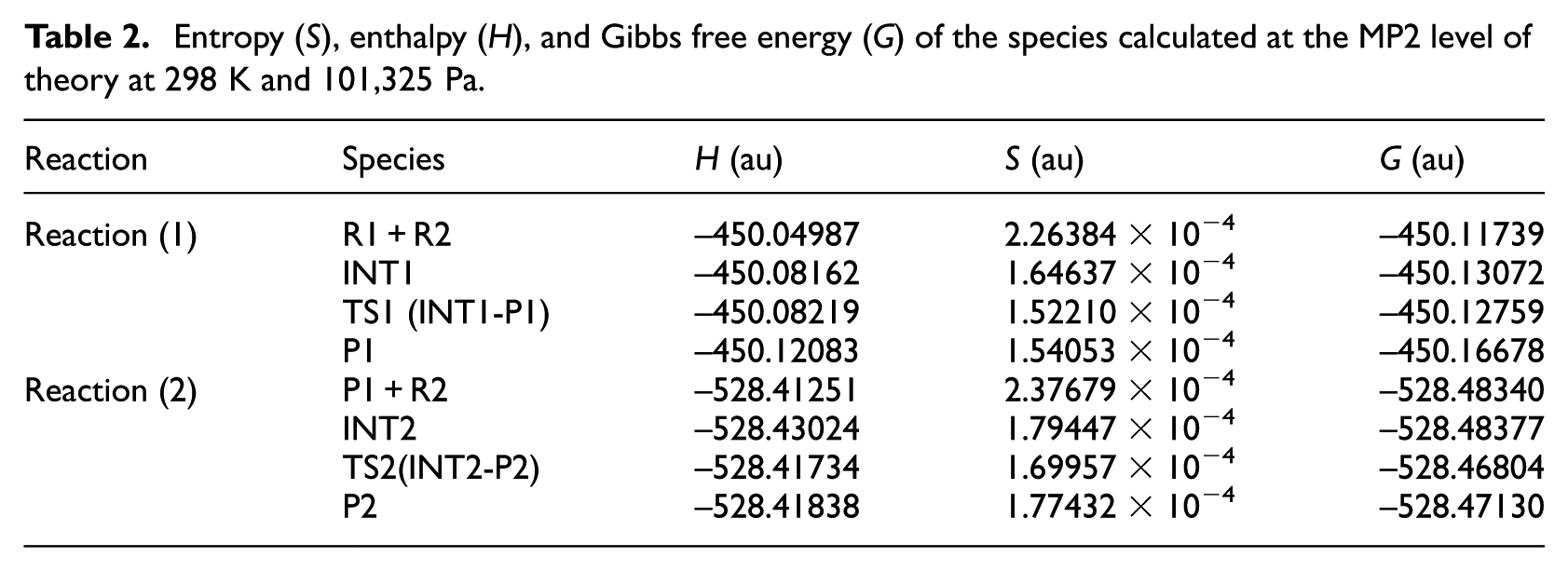
The geometrical parameters of the intermediates (INT1, INT2), transition states (TS1, TS2), and products (P1, P2) which appear in the above two reactions are given in Figure 1; the energies are listed in Table 1; the entropies, enthalpies, and Gibbs free energies are listed in Table 2; and the potential energy profiles of the above two reactions are shown in Figure 2.
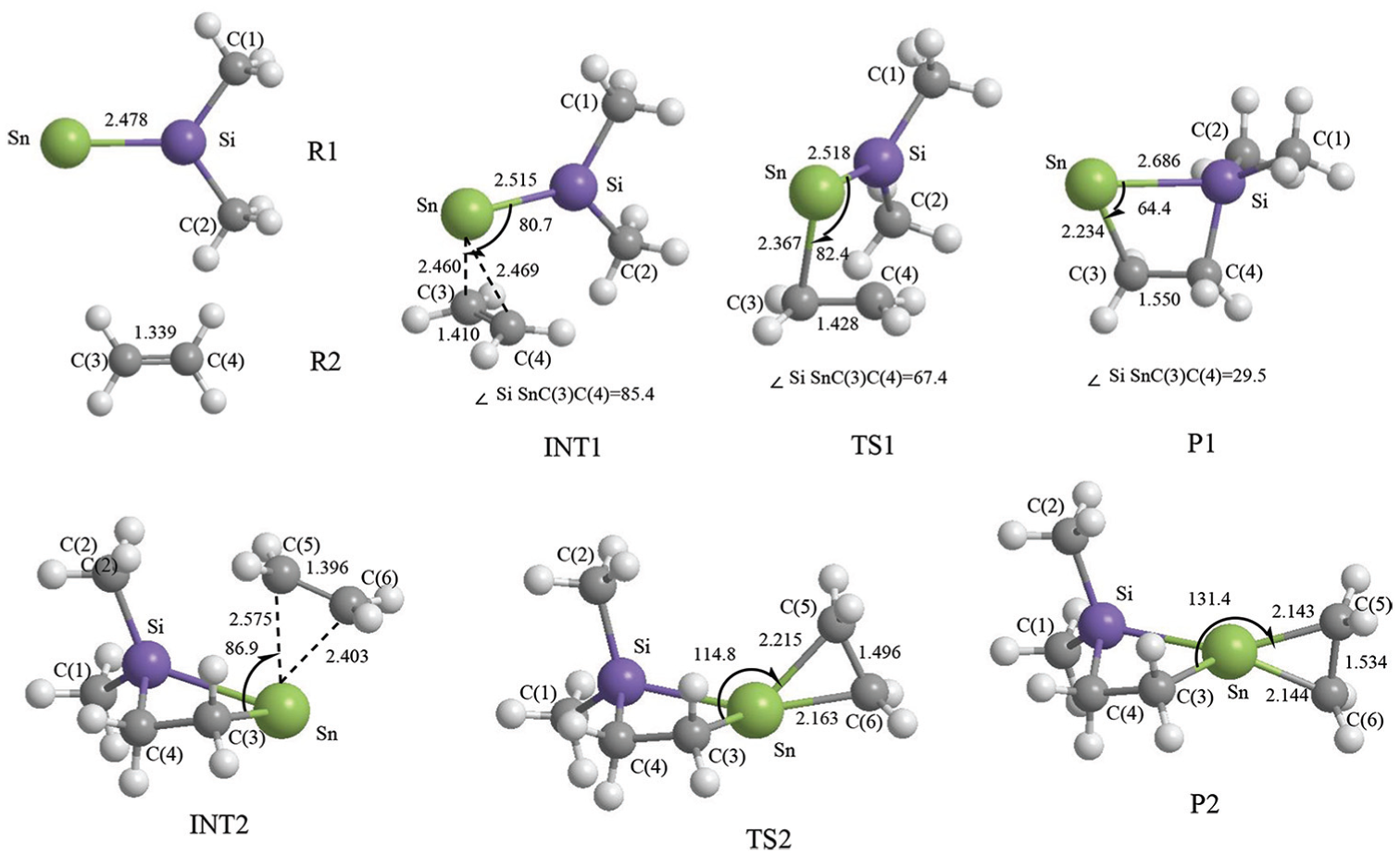
Optimized geometrical parameters and atom numbering for the species in the cycloaddition reaction between Me2Si=Sn: and ethene. Bond lengths and bond angles are in angstrom and degrees, respectively.
Electronic structure energies (Eese) and relative energies (ER) of the species calculated at the MP2 level of theory at 298 K and 101,325 Pa.
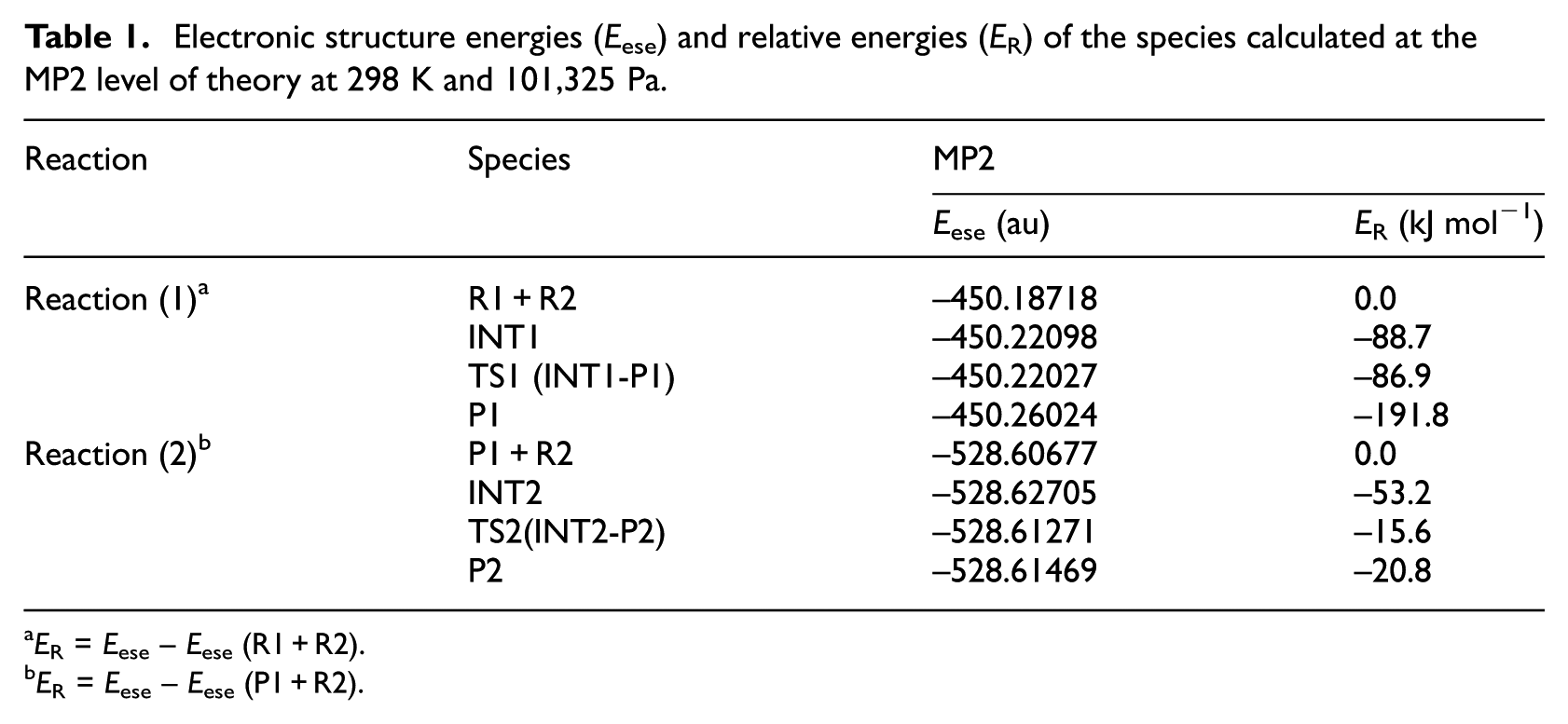
ER = Eese – Eese (R1 + R2).
E R = Eese – Eese (P1 + R2).
Entropy (S), enthalpy (H), and Gibbs free energy (G) of the species calculated at the MP2 level of theory at 298 K and 101,325 Pa.
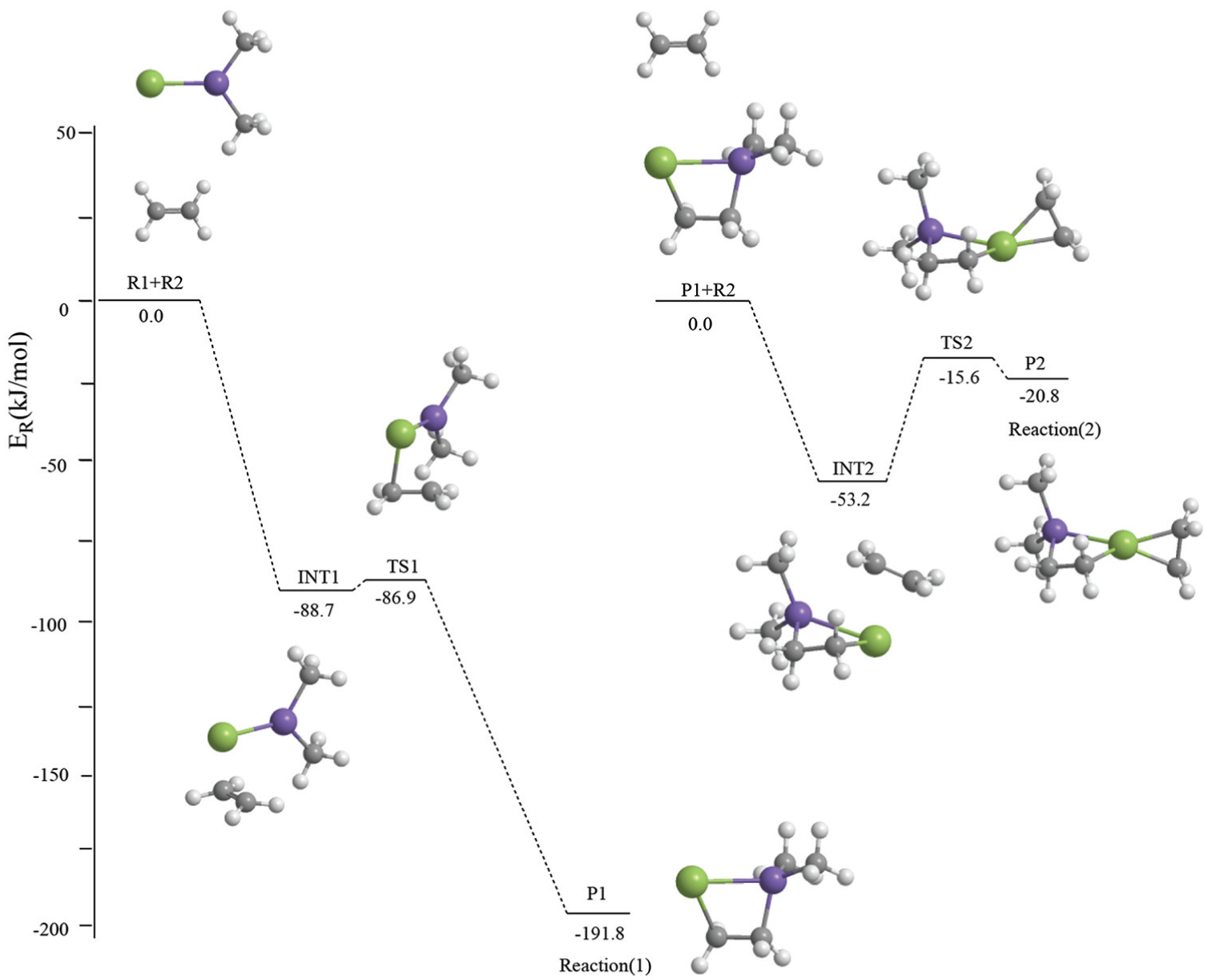
Potential energy profiles of the cycloaddition reactions between Me2Si=Sn: and ethene calculated at the MP2 level of theory.
The unique imaginary frequencies of the transition states TS1 and TS2 through vibrational analysis are 72.0i and 63.8i, respectively; therefore, these transition states can be confirmed as real ones. IRC (with the step-length of 0.1 amu−1/2 Bohr) analysis confirms that TS1 connects INT1 and P1, and TS2 connects INT2 and P2.
According to Figure 2, it can be seen that reaction (1) consists of two steps: in the first step, the two reactants (R1, R2) form an intermediate (INT1). According to Figure 2 and Table 2, the reaction is a barrier-free exothermic reaction, and the molar constant volume heat of reaction (ΔrUm) and the molar heat of reaction (ΔrHm) at normal temperature and pressure are −88.7 and −83.4 kJ mol−1, respectively, and the molar Gibbs free energy of reaction (ΔrGm) is −35.0 kJ mol−1. In the second step, INT1 isomerizes to a four-membered Si-heterocyclic ring stannylene (P1) via a transition state TS1 with an energy barrier of 1.8 kJ mol−1. According to Figure 2 and Table 2, the reaction is exothermic, and the ΔrUm and ΔrHm values at normal temperature and pressure are −103.1 and −102.9 kJ mol−1, respectively, and ΔrGm is −94.7 kJ mol−1. So, R1 + R2 → P1 will be a thermodynamically spontaneous reaction at normal temperature and pressure.
In reaction (2), the four-membered Si-heterocyclic ring stannylene (P1) further reacts with ethene (R2) to form a Si-heterocyclic spiro-Sn-heterocyclic ring compound (P2). According to Figure 2, it can be seen that the process of reaction (2) is as follows: on the basis of P1 formed in reaction (1), it further reacts with ethene (R2) to form an intermediate (INT2). According to Figure 2 and Table 2, the reaction is a barrier-free exothermic reaction, and the ΔrUm and ΔrHm values at normal temperature and pressure are −53.2 and −46.6 kJ mol−1, respectively, and ΔrGm is −1.0 kJ mol−1. Then, intermediate (INT2) isomerizes to a Si-heterocyclic spiro-Sn-heterocyclic ring compound (P2) via a transition state (TS2) with an energy barrier of 37.6 kJ mol−1. According to Figure 2 and Table 2, the reaction is endothermic, and the ΔrUm and ΔrHm values at normal temperature and pressure are 31.0 and 31.1 kJ mol−1, respectively, and ΔrGm is 32.7 kJ mol−1. Thus, the ΔrGm of INT2 → P2 is 32.7 kJ mol−1 and the ΔrGm of P1 + R2 → P2 is 31.8 kJ mol−1 and P1 + R2 → INT2 → P2 is a continuous reaction. According to the following thermodynamic equation
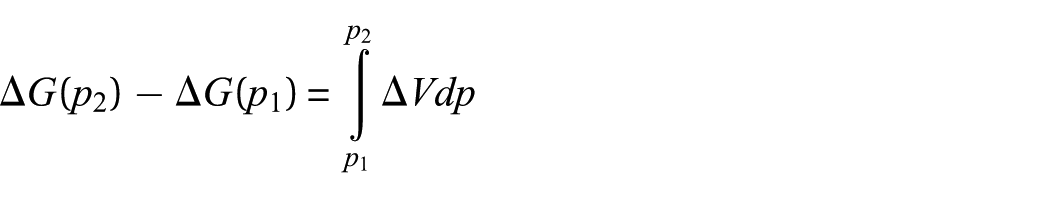
At a temperature of 298 K, the reaction P1 + R2 → P2 must proceed, and the pressure of the reaction system must be greater than 134,025 Pa (1.3 atm).
According to all analyses, reaction (2) should be the dominant reaction channel of the cycloaddition reaction between singlet Me2Si=Sn: and ethene namely
In this reaction, the frontier molecular orbitals of R1, R2, and P1 are shown in Figure 3. According to Figure 3, the mechanism of reaction (2) could be explained with frontier molecular orbital diagrams (see Figures 4 and 5). According to Figures 1 and 4, when Me2Si=Sn: (R1) initially interacts with ethene (R2), the 5p unoccupied orbital of the Sn atom in Me2Si=Sn: (R1) inserts into the π orbital of ethene to form a π → p donor–acceptor bond, leading to the formation of an intermediate (INT1). As the reaction goes on, the Sn–C(3) bond length (INT1: 2.460 Å, this is a bond length value of Sn-C(3) in INT1, TS1: 2.367 Å, P1: 2.234 Å) and the SiSnC(3)C(4) angle (INT1: 85.4°, TS1: 64.7°, P1: 29.5°) gradually decrease, and the Si–Sn and C(3)–C(4) bonds (INT1: 2.515 Å, 1.410 Å; TS1: 2.518 Å, 1.428 Å; P1: 2.686 Å, 1.550 Å) gradually lengthen. Before the transition state (TS1), Sn and C(3) form a covalent bond. After the transition state (TS1), Si and C(4) form a covalent bond. Thus, INT1 isomerizes to a four-membered Si-heterocyclic ring stannylene (P1) via transition state (TS1). Because P1 is still a reactive molecule, P1 may further react with ethene to form a Si-heterocyclic spiro-Sn-heterocyclic ring compound (P2). The mechanism of this reaction can be explained with Figures 1 and 5. According to the rule of molecular orbital symmetry, when P1 interacts with ethylene (R2), the 5p unoccupied orbital of the Sn atom in P1 inserts into the π-orbital of ethene to form a π → p donor–acceptor bond, leading to the formation of an intermediate (INT2). As the reaction goes on, the Sn–C(5) and Sn–C(6) bond lengths (INT2: 2.575 Å, 2.403 Å; TS2: 2.215 Å, 2.163 Å; P2: 2.143 Å, 2.144 Å) gradually decrease, the C(3)SnC(5) angle (INT2: 86.9°, TS2: 114.8°, P2: 131.4°) gradually increases, and the C(5)–C(6) bond (INT2: 1.396 Å, TS2: 1.496 Å, P2: 1.534 Å) gradually lengthens. Before the transition state (TS2), covalent bonds are formed between Sn and C(5), and Sn and C(6). After the transition state (TS2), the Sn atom hybridizes to a sp3 hybrid orbital. Thus, INT2 isomerizes to a Si-heterocyclic spiro-Sn-heterocyclic ring compound (P2) via transition state (TS2).
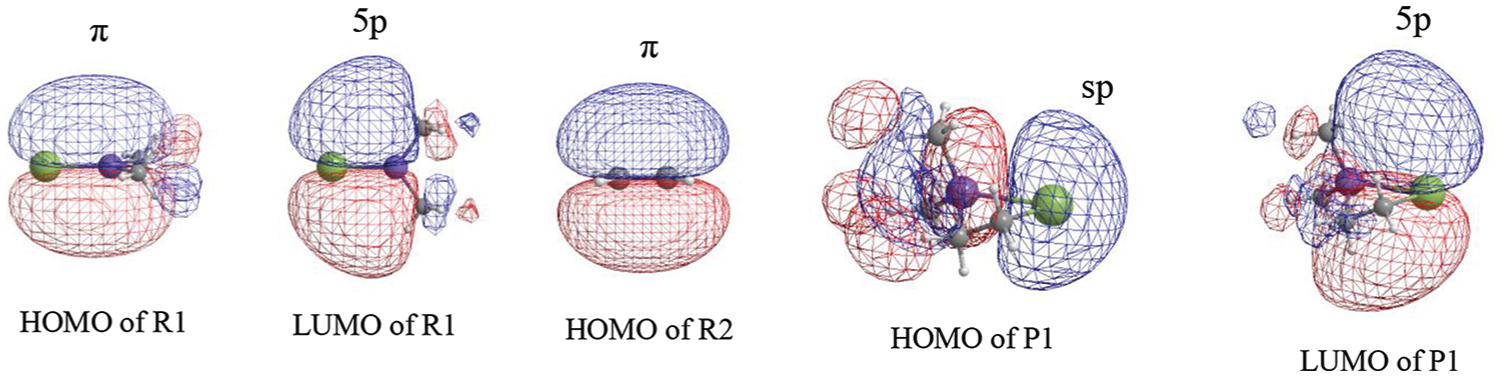
Frontier molecular orbitals of R1, R2, and P1. HOMO: Highest occupied molecular orbital; LOMO: lowest unoccupied molecular orbital.
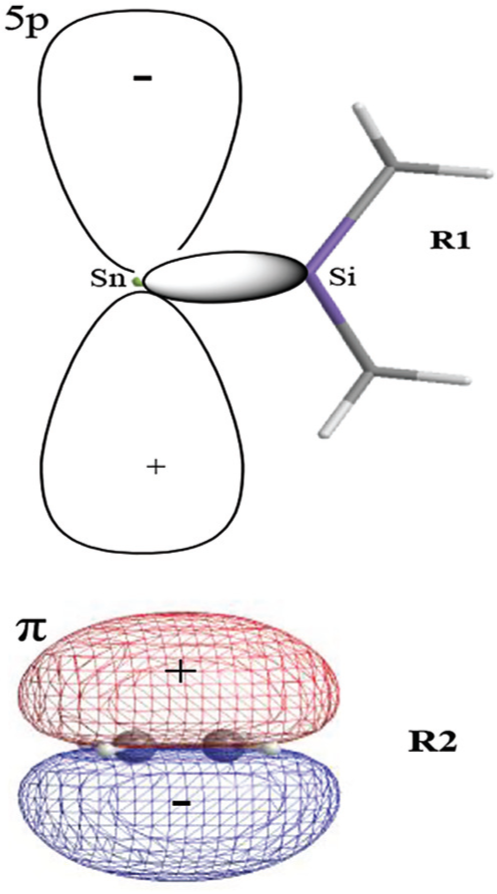
Schematic interaction diagram of the frontier orbitals of Me2Si=Sn: (R1) and C2H4 (R2).
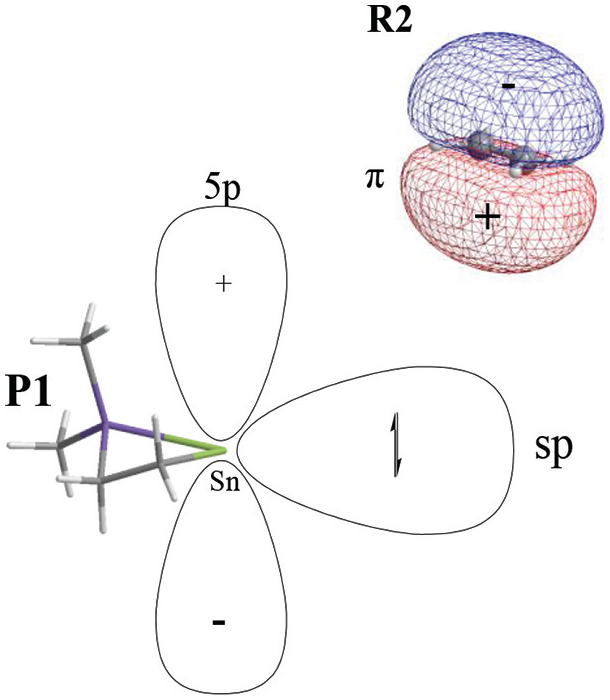
Schematic diagram of the frontier orbitals of P1 and C2H4 (R2).
Conclusion
According to the potential energy profile, the cycloaddition reaction between singlet Me2Si=Sn: and ethene obtained at the MP2 level of theory together with the 6-311++G** basis set for C, H and Si atoms and the LanL2dz basis set for Sn atoms can be predicted. This reaction has one dominant channel. This consists of four steps: (1) the two reactants first form an intermediate (INT1) through a barrier-free exothermic reaction of 88.7 kJ mol−1; (2) the intermediate (INT1) isomerizes to a four-membered Si-heterocyclic ring stannylene (P1) via transition state (TS1) with an energy barrier of 1.8 kJ mol−1; (3) the four-membered Si-heterocyclic ring stannylene (P1) further reacts with ethene (R2) to form another intermediate (INT2) through a barrier-free exothermic reaction of 53.2 kJ mol−1; and (4) the intermediate (INT2) isomerizes to a Si-heterocyclic spiro-Sn-heterocyclic ring compound (P2) via a transition state (TS2) with an energy barrier of 37.6 kJ mol−1. At a temperature of 298 K, the reaction proceeds, and the pressure of the reaction system needs to be greater than 134,025 Pa (1.3 atm).
The 5p unoccupied orbital of Sn: in X2Si=Sn: is the key aspect in the cycloaddition reaction of X2Si=Sn: and symmetric π-bonded compounds. The 5p unoccupied orbital of Sn: in X2Si=Sn: and the π orbital of symmetric π-bonded compounds form a π → p donor–acceptor bond resulting in the formation of an intermediate. The instability of the intermediate makes it isomerize to a four-membered Si-heterocyclic ring stannylene. Because the 5p unoccupied orbital of the Sn: atom in the four-membered Si-heterocyclic ring stannylene and the π-orbital of symmetric π-bonded compounds form a π → p donor–acceptor bond, the four-membered Si-heterocyclic ring stannylene further combines with the symmetric π-bonded compounds to form another intermediate. Because the Sn atom in the intermediate assumes sp3 hybridization, the intermediate isomerizes to a Si-heterocyclic spiro-Sn-heterocyclic ring compound.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.