
Abstract
Massively parallel sequencing helps create new knowledge on genes, variants and their association with disease phenotype. This important technological advancement simultaneously makes clinical decision making, using genomic information for cancer patients, more complex. Currently, identifying actionable pathogenic variants with diagnostic, prognostic, or predictive impact requires substantial manual effort.
Keywords
Introduction
Technologies such as massively parallel sequencing, also known as next-generation sequencing (NGS), allow for faster and more efficient analysis of genomes, paving the way to new knowledge by providing a large amount of data with high accuracy, in a way that is quicker and less costly than conventional sequencing techniques such as Sanger sequencing. 1 While other methodologies used for targeted analysis such as Polymerase Chain Reaction (PCR) or Quantitative PCR (qPCR) are available at lower costs, they can only detect a limited number of known variants. Highly sensitive, with a large capability for discovery, 2 NGS employment has increased with time. Sequencing data undergoes extensive processing to identify clinically relevant variants. These variants are used for disease diagnosis, prognosis, therapeutic decision, and follow-up of patients with cancer.
Lymphoma is a malignancy of the lymphatic system, a highly heterogeneous group of diseases, with more than 80 subtypes defined in recent classifications,3,4 which map to a similarly heterogeneous landscape of genetic aberrations, with hundreds of recurrently mutated genes in most entities. 5 A select number of gene variants have been identified with diagnostic, prognostic and/or predictive impact.6,7 Based on genetic alterations, improved subgrouping has been suggested in certain entities, paving the way for implementing precision medicine approaches in lymphoma. The value of NGS in lymphoma is currently being evaluated, 8 making it suitable for consideration, even if other domain restrictions, like rare diseases, would have been an alternative. While improvements have been made regarding the variant filtering process for NGS analyses, there are still variants that present difficulties in interpretation; especially structural variants, resulting in a bottleneck that requires manual work and time while still remaining error-prone.9,10 Targeted Sequencing (TS) allows researchers to use gene panels to target specific regions of interest for analysis. 11 Considering the large and diverse genomic landscape of lymphoma subtypes, it is a good candidate to apply targeted gene panels to, in order to identify the most relevant genes. The feasibility of NGS-based panel sequencing in lymphoma where results are discussed at a molecular tumour board (MTB) has also been demonstrated. 12 The use of TS here is motivated by it currently being the default method in most laboratories for the detection of somatic variants in hematological malignancies, providing a list of confirmed and potentially relevant genetic variants with diagnostic, prognostic and predictive impact. 13
An important goal of cancer care is to treat patients efficiently while minimising side effects that affect patients’ quality of life. To this aim, precision medicine will aid individual treatment decisions using targeted therapies based on genomic information and biomarkers. 14
NGS technologies have transformed genomics, but this advancement has not come without challenges in interpretation and analysis. 15 Reproducibility in results requires quality control assurance, standardized laboratory protocols for library preparation and sequencing, and effective data management and storage.16–18 This has also raised ethical, legal and societal concerns regarding privacy and informed consent.
The focus here will be on supporting clinical decision making regarding filtering variants and interpretation of remaining variants. Variants can be filtered based on their quality parameters and annotations. Filtering retains variants that reach the thresholds for different parameters such as coverage, read depth, variant allele frequency (VAF), etc. This step is usually subject to hard filtering and fixed thresholds. The variants are then filtered through a large population database to remove common variants found in the general population. Next, the remaining variants are evaluated for their association with cancer using annotations from datasets in somatic variant databases such as COSMIC. Until recently, classifications of variants for their pathogenicity lacked standardization. Each laboratory would subjectively interpret their results, leading to discrepancies and inconsistencies between reports from different labs. Two important efforts in standardisation are the ACMG/AMP guidelines for clinical classification of variants19,20 and the Belgian guidelines for biological classification of variants, 13 which categorize variants in five classes: P (pathogenic), LP (likely pathogenic), VUS (variant of uncertain significance) , LB (likely benign), B (benign).The most important advance, however, is tiering, such as the ESMO Scale for Clinical Actionability of molecular Targets (ESCAT). 21
Efforts have been made to apply AI to genomics, attempting to correlate mutations with clinical phenotypes for cancer patients. 22 With the increasing amount of available data on genes and variants detected in different diseases, it seems that AI could be a novel tool for precision hemato-oncology by providing accurate and quick diagnosis and finding the best treatment tailored to each individual, arguably shifting the definition of what constitutes a “gold standard” in cancer care.22,23 Literature databases and knowledgebases for precision oncology contain vast amounts of information on genes, diseases, biomarkers, and treatments. Exploring new and existing publications in these databases is crucial for applying the latest knowledge to research and patient care. However, this process comes at a significant cost—the time spent carefully extracting the needed information. 24 Already 20 years ago, Natural Language Processing (NLP) was combined with bioinformatics as a means to improving sequence-retrieval and sequence annotation 25 and its use of PubMed training for generative AI is currently an active research area. 26
There are several bioinformatics tools aimed at different filtering steps that can be employed but their respective performances vary widely.27,28 Hard filtering, using fixed thresholds for filtering out variants, is commonly used but adjustable thresholds may be necessary for maintaining a balance between sensitivity and specificity of variant calls. 29 This process demands extensive manual review and depends on geneticists conducting multiple assessments of the variants to draw conclusions. 30 Machine learning could streamline variant filtering and reduce the manual workload of investigating and interpreting genetic variants. Currently, the manual efforts of geneticists, who are evaluating the variants, create a significant bottleneck in delivering timely genetic analyses to clinics. While researchers have developed specific tools for various filtering tasks, few studies have examined a comprehensive, AI-augmented system to support the entire variant analysis process. 31 This gap motivates our current study.
Methods
This study was based on the Design Science Research Methodology approach (DSRM)
32
as it aims to let health informatics research and insights guide the development of a new solution by following six steps, as shown in Figure 1. For this paper, we focused on the first three steps, namely Identifying the problem, Defining objectives and Designing the solution. To this effect, a scoping review was first conducted, in February 2023, according to PRISMA-ScR
33
with details elaborated in Online Resource 1. The finely detailed outcome of the first step of DSRM, in which the problem is identified, based on all published research found relevant, is mostly in Online Resource 2. To illustrate the approach, Filtering Strategies are considered in the main text, while the supplement holds Pipelines and software, Algorithms and Artificial Intelligence. In this study, the first two steps and the beginning of the third step of the DSRM were completed.
32
For the first step, a scoping review was conducted, as well as a hands-on demonstration by two molecular geneticists, showing their current workflows. A blueprint was designed during the second step, containing the components and objectives of the proposed solution. The blueprint serves as an almost complete third step, given the validation round with expert interviews, but at the end of the third step, a prototype would also be developed and evaluated with users, left for future research.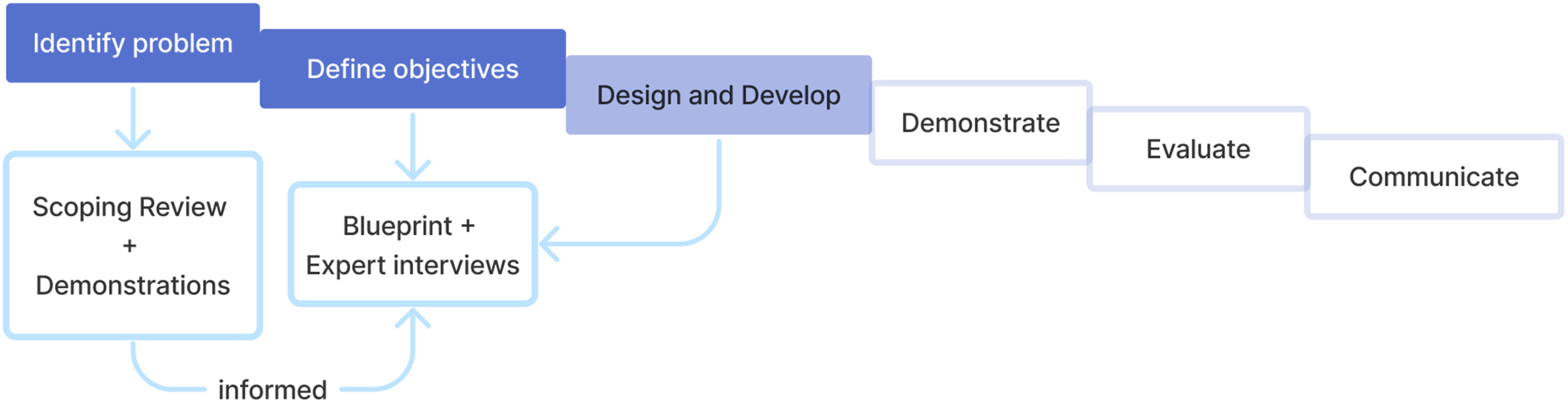
The main findings from the scoping review were used to design a solution blueprint. To gather information on the potential and pitfalls of the proposed blueprint, semi-structured interviews with experts from different fields were conducted, all of whom were stakeholders (Online Resource 3-6).
Results
DSRM1: Problem identification
Filtering strategies
Filtering strategies and criteria resulting from the scoping review.

DSRM2: Objectives
Drawing heavily on the literature findings (Table 2), the objective in the form of a blueprint is presented in Figure 2. The design starts with a pre-filtering step, which can be conducted even by existing tools. Most variant callers contain a pre-filtering option to retain only variants flagged as PASS, which pass the quality control parameters set by the caller.29,43,48 Next, the filtering process is organised in three layers: input, AI model and interface for dynamic browsing. The main sources of the input for training the model are: • a VCF file, containing a list of variants with labels confirmed by experts or in literature, • public databases for somatic variants (COSMIC), population databases containing common variants (gnomAD), and catalogues of disease-associated variants (ClinVar, HGMD), • an external NLP system for literature database mining. Literature findings that served to design the blueprint. Blueprint of an AI-augmented system for variant analysis.

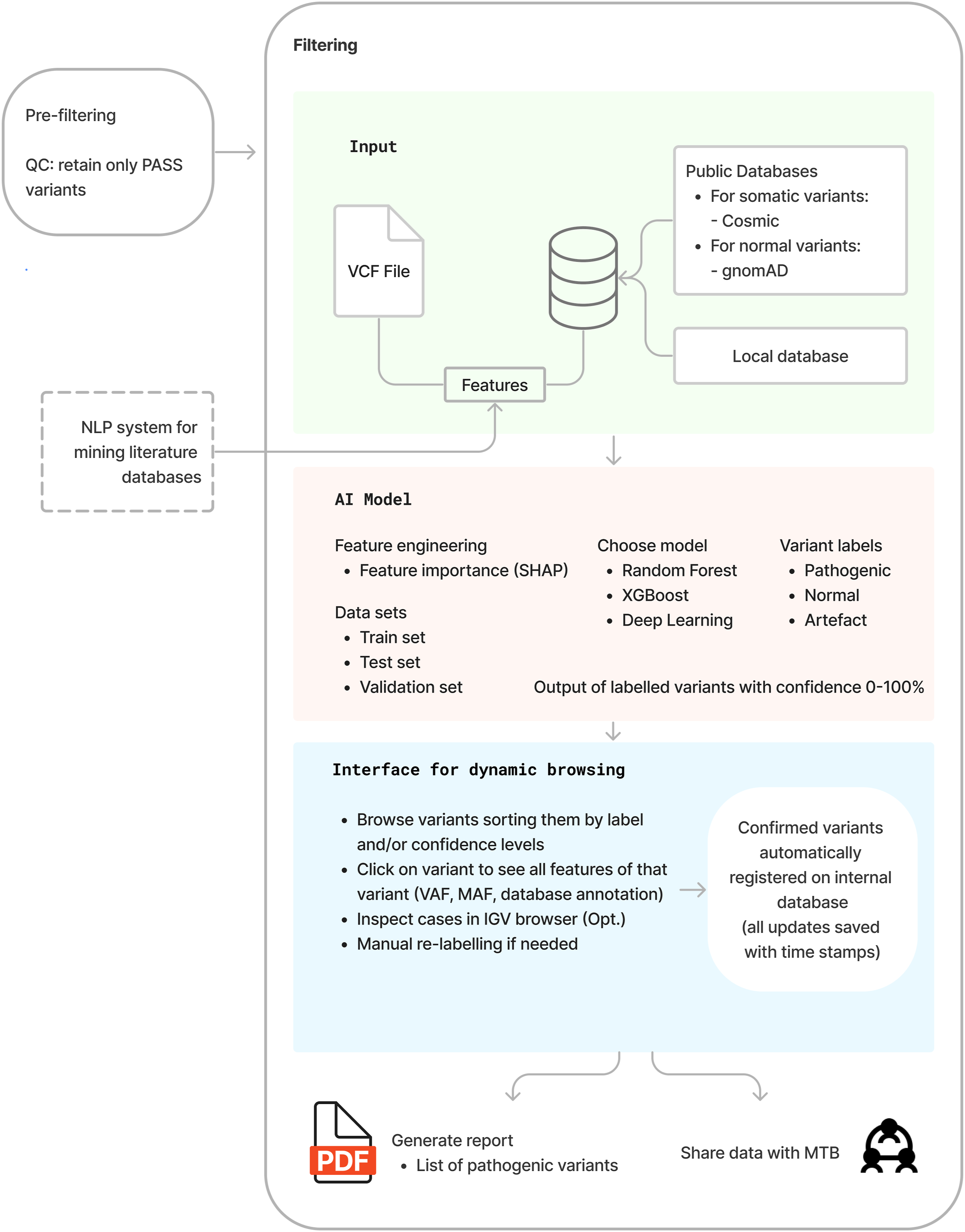
These three sources of input were used and explored to construct a combined set of features with which to build a discriminative AI model. The model should be able to find patterns in the data and avoid the need for ad hoc fixed thresholds and user-defined criteria. Firstly, feature engineering (i.e., feature selection or feature construction) is important for optimising the results of machine learning models, and using only the most informative features or combinations of features is important for scalability of the model. An exception is for deep learning models that do not require this step of feature engineering. Second, machine learning methods successfully and commonly used in relevant literature include Random Forest and XGBoost. However, other algorithms could and should be experimented with and benchmarked. For internal validation, the method of 5- or 10-fold cross validation is common practice and/or reserving 10%–20% of the data as a holdout.
59
The AI model’s output labels variants as “Pathogenic” (including P and LP), “Normal” (including VUS, LB, and B, none of which are clinically actionable), or “Artifacts.” This classification adapts to clinicians’ preference for a maximally distilled view. For research purposes, the classification task would use six classes: P, LP, VUS, B, LB, and artifact. The output labels are followed by a confidence score from zero to 100, providing a more complete view of the classification decision. Finally, an interface for dynamic browsing is necessary to visualise the results of the filtering. Geneticists should be able to browse variants by class type and confidence levels and select the ones that need closer investigation. For example, a normal variant with a very low confidence score could be selected to show the features associated with it such as VAF, MAF, annotation and if necessary, visualise it in an IGV browser to determine if it is a true variant or an artefact. The system should allow manual re-labelling and saving all labels in an internal database. At the end, a report of confirmed pathogenic variants can be generated, or alternatively, data could be sent or shared to an MTB portal, as appropriate. Experts interviewed about the blueprint solution expressed a positive attitude towards it. They provided knowledge based on their backgrounds, highlighted both challenges and opportunities, suggested involving other stakeholders, and expressed a desire for integrating multimodality into the solution, as shown in Figure 3. Attitude and knowledge elicitation are the most important and are the ones most detailed in Online Resource 6. Validation round of the blueprint with expert interviews identified five main categories and additional subcategories.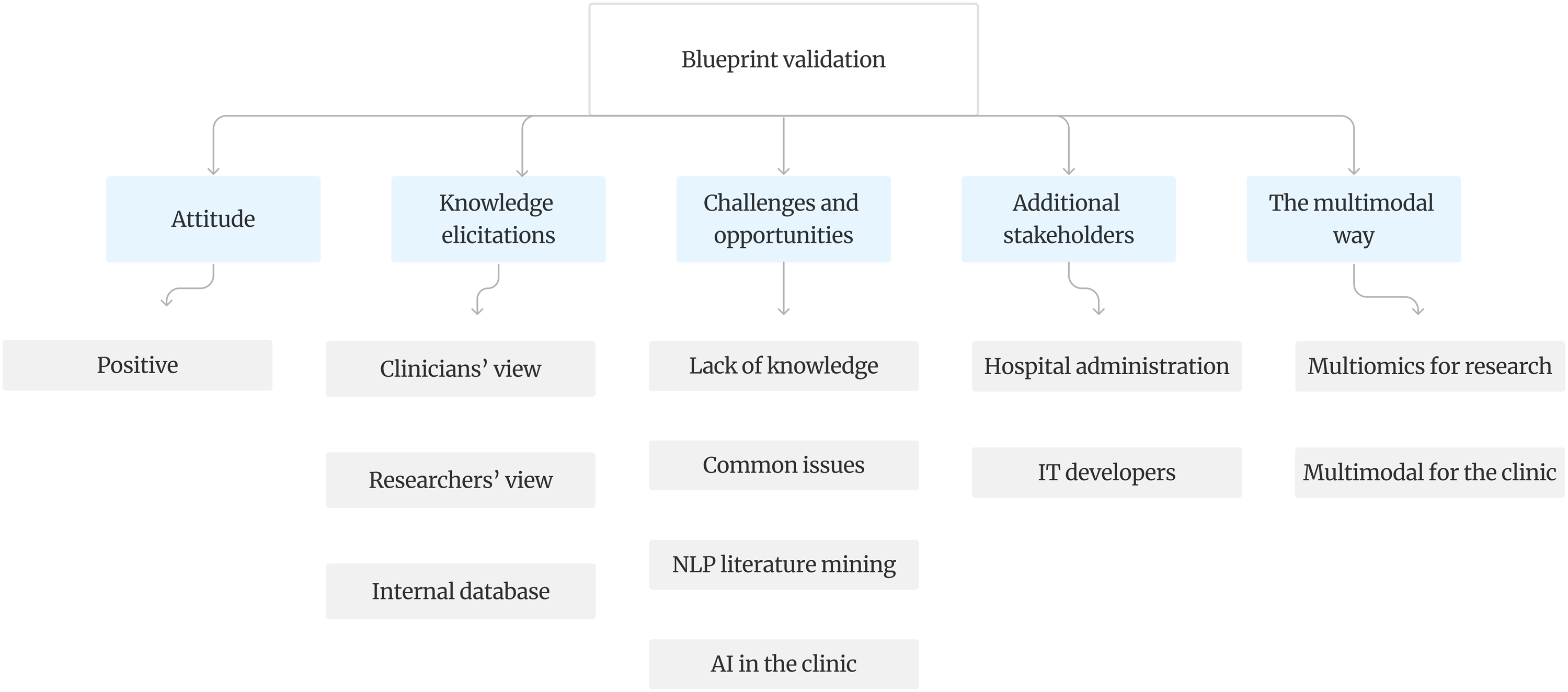
Discussion
Current challenges in variant analysis described in literature include false positives, false negatives and the presence of artefacts, each of which may result in clinical misinterpretations, 43 or even incomplete or missed diagnoses. 39 Even though several filtering tools and pipelines have been developed, the manual inspection of each candidate variant remains common practice and is time- and labour-intensive, as well as prone to error. 43 Furthermore, the variant filtering process lacks a gold standard and filtering results vary for the same sample over different platforms, different variant calling tools, 60 and even between two runs on the same platform. 28 Different strategies for variant annotation and filtering also lead to differences in the variants detected and reported. 38 In short, machine learning methods need to learn to distinguish between true variants and artefacts. Such methods can also help with interpretation of the variants using information from several layers of evidence and databases. Generative AI can be used to enable a summary of findings to be preserved alongside the supportive information from existing literature in PubMed. 26
Despite much considerations for the future of clinical genomics, the most used sequencing method today in cancer diagnostics is TS, which is best fitted for clinical use since it can benefit from highly sensitive variant calling tools focused in the regions of interest. 57 Additionally, using small-medium size panels avoids introducing large amounts of technical artefacts related with sequencing, alignment or analysis. 13 Another benefit is that TS, if it includes unique molecular identifiers and error correction, allows for the identification of variants at low VAF (as low as 0.1%–0.2%) with greater confidence. 2 Therefore, for the design of the blueprint, the focus stayed on TS and the workflows associated with it.
A system powered by machine learning methods for the filtering process has promise: eliminating hard filtering techniques would allow the model to make decisions based on patterns observed in the training data and interpret the variant by considering the complexity of combining knowledge from different features at the same time. Most tools can already today support interpretations of the variant in light of what they know about the phenotype of a particular patient. For example, consider a patient with diffuse large B-cell lymphoma and a variant in the BCL6 gene, typically expressed in the germinal centre B-cells. If histopathological examination shows aberrant expression patterns, such as BCL6 expression outside the germinal centres, it could be indicative of the functional impact of the variant. If a patient with a BCL6 variant shows significantly increased BCL-6 protein expression in the lymphoma cells compared to typical DLBCL cases, this might suggest that the variant leads to overexpression or increased stability of the BCL-6 protein. Currently, geneticists interpret the potential biological impact of most variants they find (P, LP, VUS) without knowing their true effect, especially when multiple variants are present in a single biopsy sample from tumour tissue. More functional multiomics research is needed before geneticists can draw conclusions about the actual impact on patient outcomes. As such, AI plays a crucial role when multiomics data becomes available.
Specific to lymphoma, researchers still generally lack, with some exceptions, comprehensive knowledge regarding the prognostic and treatment-decisive impact of genetic alterations. As lymphoma is so heterogeneous, this knowledge will likely need to be collected and validated in large prospective clinical trials and real-world data sets. Another promising avenue of research that could constitute a potentially clinically useful addition to guide prognosis and treatment will be longitudinal genetic analyses of circulating tumour DNA during the course of lymphoma. 61
An issue with AI models is that their performance is tested by comparing it to the ground truth, which represents expert knowledge. Studies comparing AI to clinician performance have often indicated that AI might perform as good as, or better than clinicians. 62 Therefore, if an AI model were to perform better than expert knowledge, this would not be reflected in the results, as any deviation from the human ground truth is considered as worse performance. 63 It is also a known and common occurrence for clinicians to disagree on a certain case. 64 With a great amount of inconsistencies in NGS results and reporting, even with existing standards and guidelines, it becomes a necessity to improve the consistency and replicability of NGS results. For this reason, automating parts of the process and implementing AI solutions can lead to the desired improvement of these criteria. When using databases to annotate and prioritise variants, the results will depend on the quality of the chosen databases. 65 COSMIC was chosen as the most comprehensive database of somatic cancer mutations, also supported by the scoping review, while for population databases containing common variants gnomAD was chosen, leaving out the dbSNP database as it contains a number of pathogenic cancer variants.41,66
Automatic variant analysis with interpretation by geneticists will not only reduce the time of the analysis itself but also reduce the amount of genetic data to be used for further clinical interpretation. This has implications for health informatics regarding standardisation issues when integrating NGS data with patient data, as well as implications for data visualisation, human-computer interaction and decision support when implementing variant analysis results into the clinical work process. Relevant applications include decision support for the single (hemato)oncologist at the point of care, use in MTBs to discuss single-patient cases, 12 or support of research across multiple patients and settings with clinically enriched genetic data. 67 More extensive visualisation of the integrated data in patient portals needs to be further researched.
The strengths of this study are also related to decreasing the need for manual reviewing of all filtering results from an NGS analysis. The blueprinted solution can reduce the rates of false positives and false negatives, preventing misclassification. The AI tool can also perform genetic interpretation in seconds rather than in hours, allowing for efficient scaling up in size and for the interrater variability to approach zero. Comparative studies have also shown that AI tools can achieve high concordance rates with expert panels in variant classification. Challenges include interpretability of AI predictions, securing high quality data for training, and the elaborate procedures for regulatory approval. Clinical interpretations are not explicitly dealt with, and so whether or not the analysis is linked to diagnostic, prognostic and/or predictive impact is not considered. Further development and use of the pipeline that the blueprint prescribes will take this into account. It is important to note that decision power is still with the geneticists, as they are the ones reviewing the output and examining the results more closely. 68 The automation of decisions entirely would potentially bring sequencing and its results outside the scope of prior informed consent. In contrast to published literature describing experiments, algorithms and pipelines to improve filtering process mainly through hard filtering, a systemic view has been provided that seeks to integrate all sub-processes of variant filtering. An important addition can be the incorporation in an MTB where a multidisciplinary team consisting of oncologists, pathologists, geneticists, hematologists and bioinformaticians gather to discuss findings for especially difficult and complex patient cases. 12 Not only will this solution benefit the most complex patient cases but will also bring targeted cancer care closer to every lymphoma patient by improving the efficiency and efficacy of conducting NGS research. While this study was shaped by the context of lymphoma, it is not exclusive to it. The proposed solution can be generalised to other heterogeneous cancers, including both solid and hematological malignancies.
This study has several limitations. The results of the scoping review were formulated by a single reviewer, possibly introducing bias in what was considered important to include. No librarians were employed for a more thorough search that would involve more sources than only the three main databases used in this study. Although older publications might have been interesting to examine, most of the papers included in this study mentioned the lack of a gold standard which suggests that the present choice had no severe consequences regarding missed articles because of the publication year delimiter. Snowballing did allow for finding articles that were not found by the search strategy.
Limitations of the proposed solution are mainly related to feasibility. Considering the various data sources included in the input layer, their integration and the best feature compositions will be subject to further research and experiments. Another limitation is data availability. Building such a system, where the “small n, large p” problem is a recurrent issue in genomic data, could prove to be challenging. Therefore, improving data sharing 69 between laboratories and publishing their results on genomic variants will be necessary for the proposed solution. Investigations into the link from genetic variants to phenotype were limited to improving the filtering process of NGS results. Data sharing and data availability will be even more important when dealing with linking variants to phenotype. In the case of lymphoma, an AI-powered solution would require enough representations for each of the subtypes to reach good performance in making predictions. Separating P and LP variants from the rest is desirable, as they will be important for further interpretation and represent clinically actionable variants. For the blueprint to be realised at the clinic, ethics must be considered. A principle of central importance from a legal and ethical perspective is that of autonomy and that is in its turn guaranteed by securing prior informed consent. Patients have the right to be informed of the ways data, the use of data, and data sharing could potentially impact their right to privacy. In cases where the participant will not receive the benefits of the research on the data, it is considered particularly important in a research setting to provide an understanding of the risks and benefits of participation, so that the participant may make an informed and voluntary consent. The complexity of the technology used, the potentially unexpected results it could generate as well as the difficulties in mapping the exact process steps when this is performed by means of machine learning are potentially sources of complication that need to be explored when drafting ethical guidelines.
Taking into consideration these practical limitations, it is important to identify consent for genomic analysis within consent-in-practice, and develop new forms of consensual approaches. To achieve it, one has to actively work with a reorientation of the bioethical discussion towards the under-explored area of patients’ understandings of the communicative process that is consent-in-action, instead of insisting on a strict legalistic perspective that focuses primarily on the interpretation of consent forms.
Conclusion
The results presented should not be interpreted as the final word or a definitive solution, as the entire field of variant analysis and interpretation is under rapid methodological development.70,71 This makes blueprints and process flow charts tentative, in anticipation of clinical trials. With that in mind, this study was able to propose a design of an AI-augmented system for variant filtering and analysis, which would reduce the costly and error-prone manual labour associated with this process. There are many possibilities for further research.
Supplemental Material
Supplemental Material - Smart variant filtering - A blueprint solution for massively parallel sequencing-based variant analysis
Supplemental Material for Smart variant filtering - A blueprint solution for massively parallel sequencing-based variant analysis by Orlinda Brahimllari, Sandra Eloranta, Patrik Georgii-Hemming, Zahra Haider, Sabine Koch, Aleksandra Krstic, Frantzeska Papadopoulou Skarp, Richard Rosenquist, Karin E. Smedby, Fulya Taylan, Birna Thorvaldsdottir, Valtteri Wirta, Tove Wästerlid, Magnus Boman in Health Informatics Journal.
Footnotes
Acknowledgment
The authors thank Noura Ezaz-Nikpay for comments on an earlier draft.
Declaration of conflicting interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: RR received honoraria from Abbvie, AstraZeneca, Janssen, Illumina and Roche.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Vetenskapsradet; 2021-04610.
Ethical statement
Data availability statement
No datasets were created or stored within this study.
Supplemental Material
Supplemental material for this article is available online. Auxiliary tables and figures have been deposited as online resources.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.