
Abstract
Background:
Carpal tunnel syndrome (CTS) is the most common entrapment neuropathy of the upper extremity, and its surgical treatment is carpal tunnel release (CTR). It is mostly performed in local anesthesia. There are no clinical randomized controlled trials (RCTs) comparing local infiltration anesthesia with or without a distal median nerve block in CTR. The aim of the PERSONNEL trial (carPal tunnEl ReleaSe lOcal iNfiltratioN mEdian bLockade) is to assess whether a distal median nerve block reduces pain during and after the procedure in addition to local anesthesia.
Methods:
This is a single-center randomized clinical superiority trial comparing local anesthesia alone and local anesthesia with a distal median nerve block for CTR in patients with CTS. Adult patients will be randomized in one university hospital in Finland, and the intended sample size is 118. The primary outcome is the mean pain level after the procedure during 72 h using a visual analogue scale (VAS). The secondary outcomes include expected pain; pain during the injection of the anesthetic solution caused by pressure, burning, needle sting, and total pain; worst pain during the surgery; duration of anesthesia; number of experienced needle stings; Boston Carpal Tunnel Syndrome Questionnaire (BCTSQ); pain killer consumption;, patient satisfaction using Net Promoter Score (NPS); and complications.
Discussion:
Patient satisfaction is a crucial factor in modern healthcare. A distal median nerve block may reduce pain during and after CTR, potentially increasing patient satisfaction with the given treatment. It can also be hypothesized that better postoperative pain control may prevent complications, for example, complex regional pain syndrome. However, we lack adequate evidence to justify the use of distal median nerve block, which can itself predispose patients to complications, for example, median nerve injury. Therefore, there is a need for adequate RCTs to assess its efficacy. The results of this study can be used to optimize anesthesia for carpal tunnel surgery, improve patient satisfaction, and possibly prevent complications.
Registration:
ClinicalTrials.gov NCT05372393.
Keywords
Context and Relevance
This is a trial protocol for a randomized clinical trial to compare local anesthesia with or without distal median nerve block. To the best of our knowledge, this is the first RCT on this topic. Although this procedure is performed often, its benefits are unknown, and it carries risks, for example, median nerve injury. The aim of this trial is to assess whether adding a distal median nerve block to local anesthesia reduces the patient’s perceived pain level compared with using only local anesthesia. The null hypothesis is that the use of distal median nerve block with local anesthesia does not reduce pain after CTR compared with local anesthesia alone. There is a need to assess if the use of distal median nerve block is justified. The results of this trial can be used to optimize anesthesia for carpal tunnel surgery, improve patient satisfaction, and possibly prevent complications.
Background
Carpal tunnel syndrome (CTS) is the most common entrapment neuropathy of the upper extremity, with an increasing prevalence and severity with age.1,2 Adults between 40 and 60 years constitute the largest patient group seeking treatment for CTS. 3 CTS has a multifactorial etiology; however, in many cases, it is classified as idiopathic. 4 Women develop CTS three times more likely than men. 2 Other risk factors include diabetes mellitus, 5 hypothyroidism, 6 obesity, 7 rheumatoid arthritis, 8 menopause, 9 and pregnancy. 10 Manual labor that requires repetitive movements, force, or the use of hand-operated vibratory tools is associated with a high risk of CTS. 11
When the pressure increases in the carpal tunnel, it leads to compression, ischemia, and eventually damage to the sensory and motor fibers of the median nerve, causing the typical symptoms,12,13 including pain, 4 paraesthesia, 4 and tingling. 14 Difficulty in holding objects is a late symptom caused by the affection of motor fibers. 1
Anamnesis plays a significant role in raising suspicion of CTS. Clinical examination is key to diagnosis, with cornerstone tests including median nerve compression tests, for example, Phalen and Durgan. 15 Nerve conduction studies can be used to confirm the diagnosis. 1
Treatment options include both non-operative and operative methods. Non-operative treatment is recommended in mild to moderate cases, whereas proceeding directly to operative decompression is advocated in severe cases judged by clinical examination and nerve conduction studies.1,14 Non-operative treatment includes, for example, splinting, 16 corticosteroid injection, 16 and physical therapy. 17 Physical therapy can include, for example, therapeutic exercises and manipulations and ultrasound. Surgical treatment (carpal tunnel release [CTR]) involves division of the transverse carpal ligament. CTR is performed either open, mini-open, or with an endoscope. 18 Surgery can be performed under the axillary or intravenous block or general anesthesia, but recently Wide-Awake Local Anesthesia with No Tourniquet (WALANT) has gained more popularity.
Rationale of the trial
Various methods have been used to reduce pain during CTR, including local anesthesia without a tourniquet. 19 Nerve blocks alone at the wrist have been proven to provide effective anesthesia for CTR. 20
There are no prior randomized controlled trials (RCTs) comparing local infiltration anesthesia to local infiltration anesthesia augmented with a distal median nerve block in CTR. Distal median block in CTR is believed to reduce pain intra- and postoperatively. However, the superiority of distal median block with local anesthesia compared with pure local anesthesia alone has not been proven.
Trial aim and hypothesis
The aim of this trial is to assess whether adding a distal median nerve block to local anesthesia reduces the patient’s perceived pain level for up to 72 hours after CTR, compared to using only local anesthesia—an anesthesia mixture injected solely in and around the planned incision and nerve release.
Methods
This trial protocol is reported in accordance with the “Standard Protocol Items: Recommendations for Interventional Trials (SPIRIT) Statement” (Supplementary material, Supplementary Appendix I). 21
Study design
PERSONNEL (Carpal Tunnel Release Local Infiltration Median Blockade) is a double-blind, superiority randomized controlled clinical trial in patients with CTS, where both the patients and investigators are blinded. superiority randomized controlled clinical trial in patients with CTS. Patients will be randomly divided into two parallel trial groups, 1:1 in size to each other.
Patient selection and diagnostics
Inclusion and exclusion criteria
Adult patients diagnosed with treatment-naive CTS, without factors that could distort the results or present severe risks for surgery, will be recruited for the trial (Table 1).
Inclusion and exclusion criteria.

CTS, carpal tunnel syndrome.
Excluded patients
During recruitment, patients who meet one or more of the predefined exclusion criteria will be excluded from the study.
Randomization and blinding
Investigator AR will generate the randomization lists. AR will not take part in the treatment or recruitment of the patients and will not know which anesthetic method is used in each group. Investigator or research nurse (recruiters) will recruit the patients. The recruiter fills out identification and screening logs and calculates the patient’s Pain Catastrophizing Scale (PCS) score after the patient has completed the PCS-FINv2.0 form. We defined the cutoff value for low and high scores to be 13. Subsequently, the recruiter informs the randomization nurse about whether the patient’s treated hand is dominant or non-dominant, as well as whether the PCS score is low or high, which are the stratification criteria for this trial. CTR on the dominant hand may cause more pain after surgery since the patients may potentially use more of their dominant than the non-dominant hand even if the hand has been operated on. The PCS is used to assess pain tolerance, and our objective is to consider its implications in the context of different groups. The purpose of the stratification criteria is to ensure that the groups are balanced in terms of dominant and non-dominant hands, as well as low and high PCS scores. Both stratification criteria are prognostic, meaning that they are associated with the endpoint.
The recruiter gives the identification number of the patient for the randomization nurse who informs the allocation (purely local or local with median nerve blockade) from the previously made randomization list for the recruiter via phone. The recruiter informs caregivers which group the patient belongs to. The recruiter has identification lists that contain the patient identification number. Patients’ visibility to the anesthesia and surgery area is blocked by drapes.
This trial is double-blind, meaning that both patients and investigators are unaware of the type of anesthesia administered. Investigators remain blinded to the data and results during the data analysis and interpretation phase.
There is no need for unblinding or revealing a participant’s allocated intervention during the trial since the operation performed, anesthetic solutions, and methods are widely in use and proven safe, so there will not be unexpected adverse events that would demand revealing the blinding.
Sample size calculation
Sample size calculation is based on an assumed group mean difference of 1 cm in the postoperative 10 cm visual analogue scale (VAS) scale, with the minimal important difference MID likely being around 1.4 cm. 22 This is the difference between the groups that would not like to be missed. A common standard deviation (SD) of 1.9 cm is assumed. Based on these values, 59 patients are needed per group, for a total of 118 patients, to achieve a power of 80% and a type I error rate of 5%. Minimal dropouts are expected, and this is hence our final sample size.
Study setting and feasibility
The trial is performed at Kuopio University Hospital in Finland. Patients are recruited from the outpatient clinic of hand surgery, where they are referred for CTR. Specializing physician of hand surgery or orthopedics, or a consultant in hand surgery, will perform all the operations. Nurses will act as an assistant. Before initiating the trial, approval was acquired from The Research Ethics Committee of the Northern Savo Hospital District. The trial is registered in ClinicalTrials.gov. Recruiters will procure written informed consent from each patient. Patient information and consent forms are formulated as clearly as possible to provide every patient with understandable and adequate information about the trial. All the patients’ personal data are pseudonymized. Only essential personal data for the trial is stored in the trial register.
Interventions
CTR is performed under WALANT. The first group will have a pure local, and the second a distal median nerve block with local infiltration anesthesia. For both groups, the anesthetic cocktail consists of 1 ml of (7.5%) sodium bicarbonate (Natriumbicarbonate Braun 75 mg/ml), 4.5 ml of lidocaine with adrenaline 1% (Lidocaine cum adrenalin 10 mg/ml), and 4.5 ml of bupivacaine with adrenaline 0.5% (Marcain cum adrenalin 5 mg/ml + 5 µg/ml). The anesthetic cocktail must be prepared in the above-mentioned order to avoid possible precipitation. A 24-gauge hypodermic needle and 10-ml syringe are used to inject the solution. For the distal median nerve block group, half of the anesthetic solution is injected into the median nerve area 5 to 7 cm proximally from the distal wrist crease (Fig. 1). The other half is injected locally. In the purely local anesthesia group, the care provider pinches from the area of median nerve blockade prior to performing local anesthesia. The goal of pinching is to mimic the injection of the distal median nerve block and to enhance the blinding of the patients. All the anesthetic solution is injected locally in the purely local anesthesia group. In both groups, additional anesthetic solution can be injected locally if necessary.
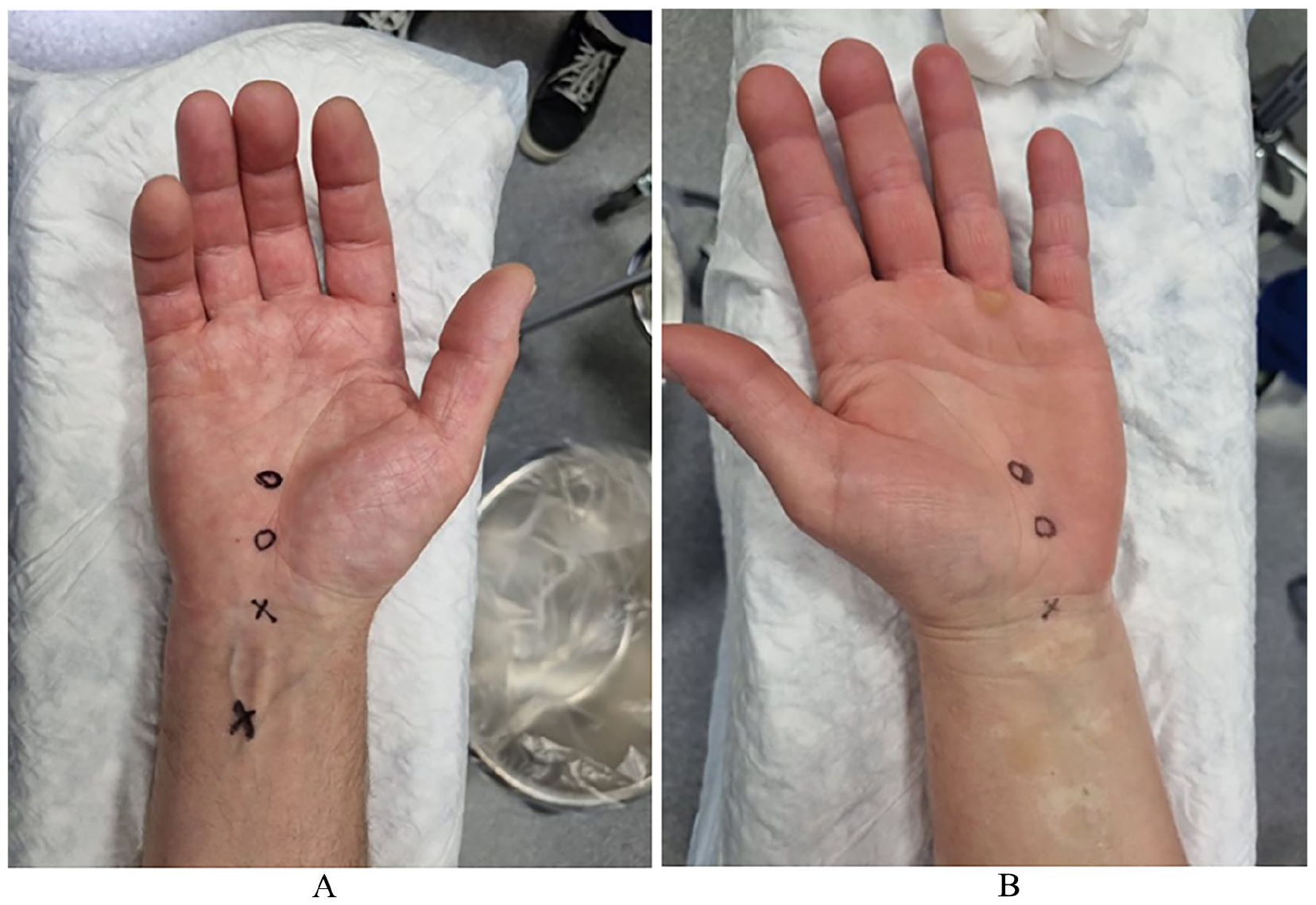
(A) Injection of local anesthesia and median nerve block. (B) Injection of local anesthesia.
The transverse carpal ligament will be divided longitudinally under direct vision until the median nerve is fully decompressed. The incision is closed with sutures. The wound is covered with a light hand surgical dressing for a few days. For postoperative pain management, patients will be prescribed paracetamol and ibuprofen. Patients will be given self-care instructions.
Follow-up
Postoperative follow-up data will be gathered by General Data Protection Regulation (GPDR) approved survey software, SurveyMonkey (Momentive, San Mateo, California, USA). Before the operation, all the patients will be given instructions for the follow-up surveys and questionnaires. A link to postoperative online forms will be sent to patients via email immediately after the operation and 3 months later. Perceived pain is measured until the third night postoperatively with an electric form that participants must fill every fourth hour after the procedure, excluding nights, and estimate pain level by using a visual analogue scale (VAS). The consumption of painkillers is measured during the first 72 h postoperatively. The duration of anesthesia is registered by asking the patients to fill out the online form when they first time feel pain in the operation field or have to use painkillers. After 3 months of the operation, the Boston Carpal Tunnel Syndrome Questionnaire (BCTQ) score is used again to determine patients’ symptom severity and overall functional status to compare the results with preoperative estimations. Using Net Promoter Score (NPS), the patients are asked to evaluate how likely they would recommend the procedure. In addition, patients are asked if they have had any adverse events during the last 3 months possibly caused by the procedure.
The patients will be contacted by email or phone if they have not filled out the surveys or questionnaires within 2 weeks from the follow-up time point (3 months). If necessary, the surveys or questionnaires will be filled in by phone.
Outcome parameters and statistical analysis
Primary outcome
The primary outcome is the pain level perceived by the patient after the procedure during the first 72 h using the VAS. The VAS is a unidimensional pain-rating scale, a straight line in which one end means no pain and the other the worst pain that can be imagined. The use of VAS is simple for both patients and recruiters. VAS is assessed every fourth hour while awake. The first assessment is performed 4 h after the surgery. The mean VAS is calculated from all time points over the 72-h period. We will evaluate the clinical significance of the VAS result. MID should be more than 14 mm, 22 and if the mean difference exceeds this threshold, the comparative treatment can be found to be more effective than the control. VAS is a reliable and valid pain-rating scale. 23
Secondary outcomes
Secondary outcomes include patient-rated outcome measures, safety, the entire consumption of painkillers after the surgery during the first 72 h postoperatively, pain of performing the anesthesia, and pain during and after the operation (Table 2).
Primary and secondary outcomes and their assessment points.
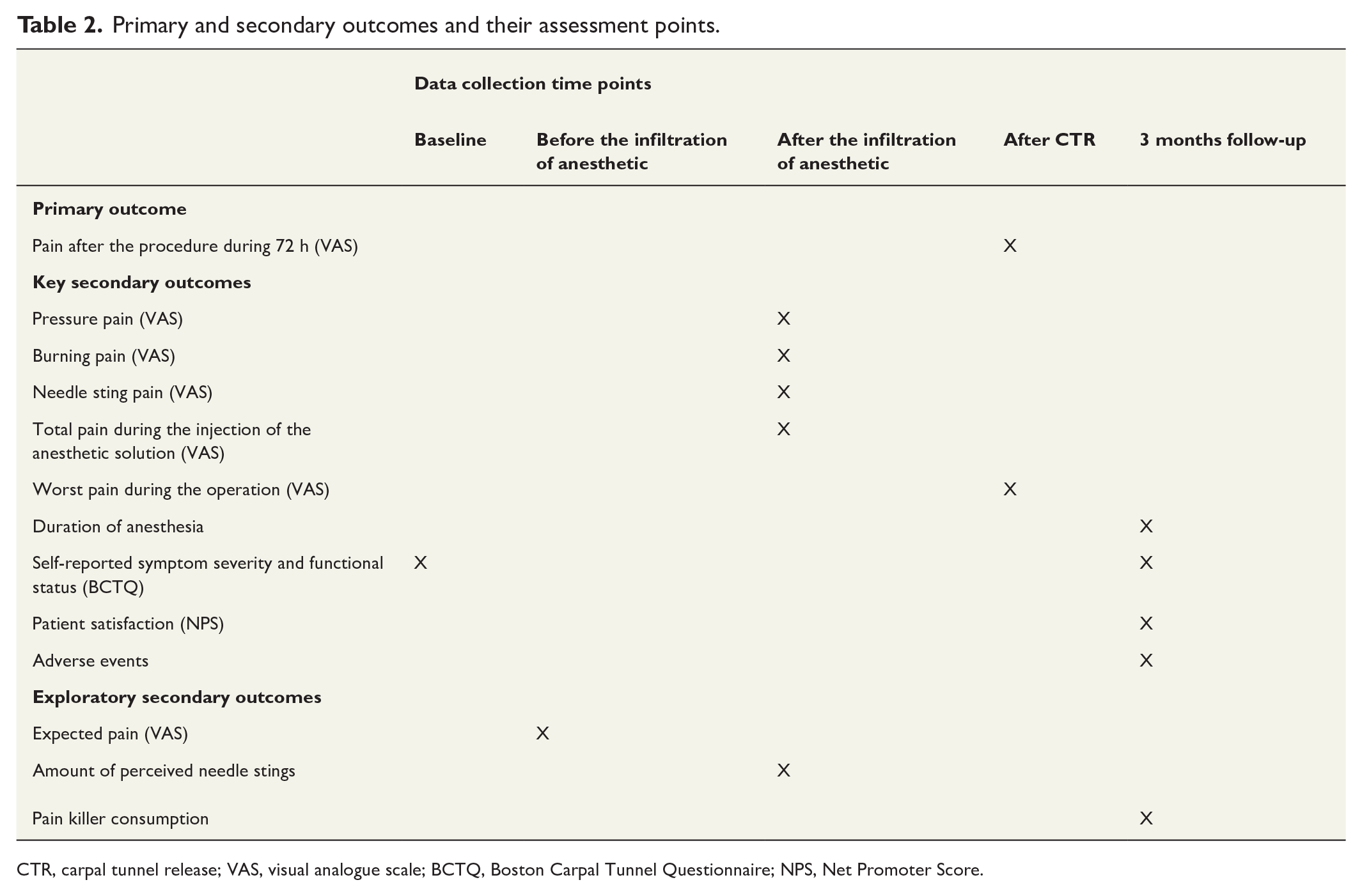
CTR, carpal tunnel release; VAS, visual analogue scale; BCTQ, Boston Carpal Tunnel Questionnaire; NPS, Net Promoter Score.
Statistical hypothesis
The primary objective of this RCT is to compare pure local anesthesia to local anesthesia with distal median nerve block in patients undergoing CTR. The null hypothesis is that the use of distal median nerve block with local anesthesia does not reduce pain after CTR compared with pure local anesthesia. Conversely, the alternative hypothesis is that a significant difference exists between the two groups in the pain level perceived by the patient after the procedure during the first 72 h using the VAS. Secondary hypotheses will explore potential differences in secondary outcomes; including pressure, burning, needle sting and total pain during the injection of the anesthetic solution; worst pain during the operation; duration of anesthesia; self-reported symptom severity and functional status (BCTQ); patient satisfaction (NPS); adverse events; expected pain; amount of perceived needle stings; and pain killer consumption.
Data analysis plan
Patients will receive a cover letter, patient information leaflet, and consent form along with the invitation letter for the CTR, allowing them to familiarize themselves with the documents in advance. Recruitment will occur on the day of the operation. The recruiters enroll patients, and if necessary, they contact the primary junior investigator if they are unable to answer the patients’ questions. If the patient consents to the trial, the recruiter obtains written consent and collects the baseline data prior to randomization. The baseline data includes age, gender, chronic diseases, continuously used painkillers, history of smoking, self-reported symptom severity and functional status, expected pain during the injection of the anesthetic solution, and PCS Score. After 3 months of operation, patients must fill out the online trial forms for postoperative follow-up (Fig. 2). All the follow-up is performed in SurveyMonkey, so there will be no additional visits for patients due to the research.
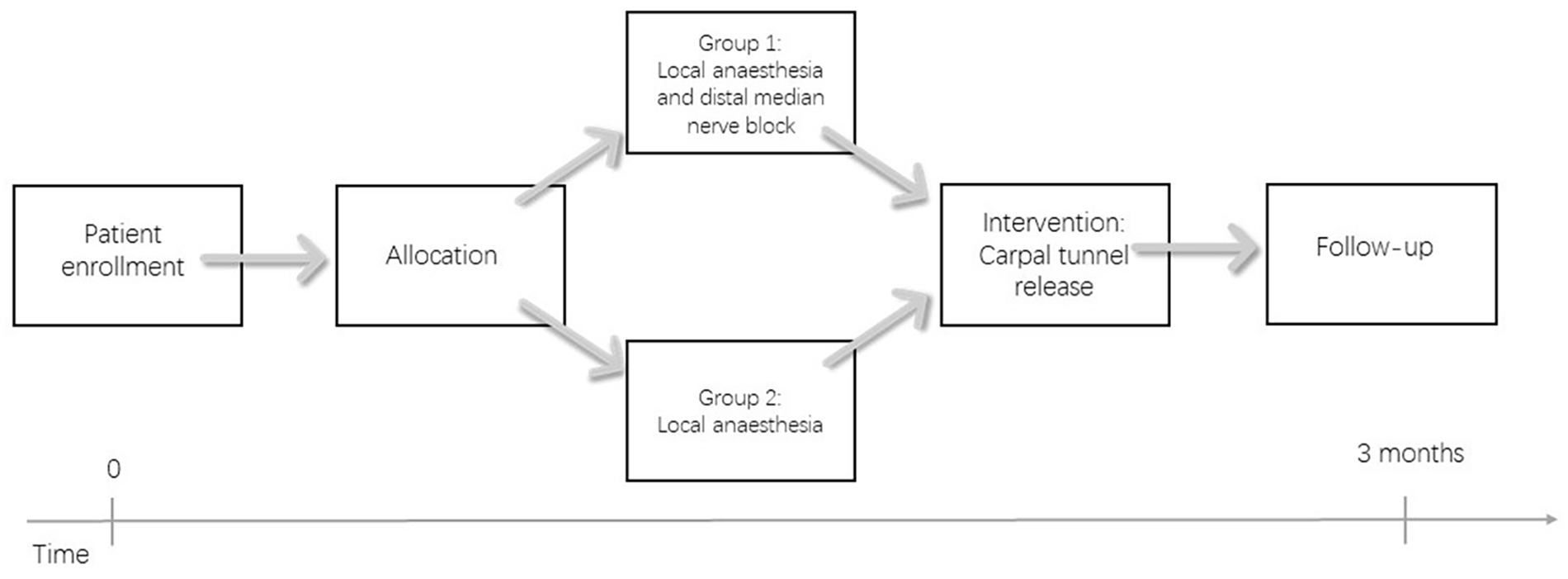
Flow chart of the trial.
All the patients’ personal data are pseudonymized. Patient’s personal data cannot be connected to the patient without additional information. The patient’s running number is found in the patient system and a separate log.
Data are collected peri- and postoperatively using trial forms. Preoperatively, the patient is asked to fill out the BCTQ and the PCS questionnaire score form (PCS-FINv2.0). BCTQ determines symptom severity and overall functional status. Expected pain during the injection of the anesthetic solution is estimated before the anesthesia is performed by using the VAS scale. Immediately after the infiltration of the anesthetic solution, patients will be asked how many needle stings they felt when the anesthesia was performed. In addition, using the VAS scale, the pain perceived during the injection of the anesthetic solution caused by the needle sting, administration of the anesthetic solution (tension- and burning), and total pain caused by injection will be measured. Immediately after the operation, patients will be asked what level the worst pain was on the VAS scale during the operation.
To minimize any bias in interpreting the findings, AR, who will be blinded for the treatment allocation, performs data analysis. AR will present the results blinded (groups A and B) to the writing committee, and the committee will reach a consensus on the interpretation of the findings. After consensus, the groups will be unblinded. 24
The descriptive statistics will be presented as means with SD, as medians with interquartile range, or as counts with percentages. Linear regression is used to estimate the mean difference in continuous outcome measures between treatment groups. For the primary analysis, a multivariable model is prepared which includes the treated group as a binary variable, patient-reported pain as an outcome, and PCS, age, side, and gender as covariates. As a result, there will be an adjusted difference between the groups regarding the treatment options. Similar analyses are performed for secondary outcomes using the same methods, except for BCTQ, in which the baseline is added as a confounding factor using a multivariate model.
The statistical analysis is performed using RStudio version 4.
Interim analysis, stopping guidelines, and committees
In this trial, no interim analyses will be performed, and no stopping guidelines are needed since the studied treatments are widely used and shown to be safe. This trial does not have an endpoint adjudication committee or a data management and safety committee for similar reasons. The writing committee includes all the authors of this protocol, and the steering committee consists of the senior investigators of the trial (YN, AR, JS, and MPR).
We collect data on all treatment-related complications and report them (Table 3). In the case of a serious adverse event, it will be treated in accordance with prevailing treatment practices. Adverse events are treated similarly to any patient regardless of the trial. CTR and the anesthetic methods used in the trial are well-known, commonly used, and found to be safe, so the expected value is that the risk of serious adverse events is low and is equivalent to normal treatment. Since the trial serves routine treatment methods, possible treatment injuries will be compensated by the Finnish Patient Insurance Center.
Possible complications from performing anesthesia and carpal tunnel release, defined by the Clavien-Dindo classification.

CNS, central nervous system; IC, intensive care; ICU, intensive care unit.
Withdrawal
The duration of the follow-up in the trial is short; hence, there will not be a significant number of dropouts. Junior investigators and the research nurse will actively monitor that the patients return the follow-up questionnaires, and if they are missing, patients are contacted by email or telephone. Treatments studied in this trial are widely used and proven to work and to be safe, so there will not be a need to discontinue or modify allocated interventions. Patients have the right to withdraw from the trial at any point without providing a reason, and the collected data will be used as part of the trial data.
Dissemination and ethics
The trial’s registrar is the Northern Savo Hospital District, responsible for the legality of personal data processing of the trial. Only essential personal data for the trial is stored in the trial register, for example, neither names nor social security numbers will be saved. All confidential personal patient information is securely kept in a locked cabinet in the research nurse’s office. Only research personnel involved in the trial, including the research nurse, data analysts, and investigators, will have access to the collected data. Data will not be shared outside the EU/ETA area. As this is a clinical trial, the data will be stored for 15 years, encompassing the time from the trial’s commencement to the end of data storage. After 15 years, the data will be securely destroyed. If a patient discontinues participation or withdraws consent, the collected data from the patient will be used as part of the trial data. There is no intention to use professional writers.
The protocol will be available after publication from the journal in accordance with its rules. The participant-level dataset and statistical code will be available after the trial is completed in accordance with GPDR (Supplementary Appendix II). Results of the trial will be published in a peer-reviewed journal.
If there is a need for protocol amendments, they will be informed to and accepted by the Research Ethics Committee, communicated to trial participants if necessary, recorded on ClinicalTrials.gov, and reported in the publication.
The trial has no financial interests.
Discussion
The trial aims to determine whether a distal median nerve block, in addition to local anesthesia, reduces the pain perceived by patients during and after CTR compared with using only local anesthesia. The primary outcome is postoperative pain experienced by the patient, with the primary time point being the mean value of the first 72 h after the operation. CTS is the most common entrapment neuropathy of the upper extremity,1,2 but the optimal method of anesthesia for CTR is still not yet clear.
No trial comparing local anesthesia to local anesthesia augmented with distal median nerve block has been published before. There is also no trial noting individual tolerance to pain. The quality of the median nerve block at the wrist has been achieved by using sensory or sensory-motor nerve stimulation and has been proven effective. 20 This increases trust in the effectiveness of the treatment method, but it still needs to be adequately proven which is the goal of this trial.
A strength of the trial lies in its loose inclusion and exclusion criteria, facilitating results that can be generalized to typical surgical treatments for CTS. A limitation of the trial is that pain tolerance is partly cultural and depends partly on cultural features, potentially restricting the global validity of the results. Another limitation is that blinding of care providers (surgeon and the procedural room nurses) is not reasoned in this trial. While there is a risk that a complete distal median nerve block may not be achieved due to the procedure being performed without sonography; we do not consider it as a limitation. This is because our trial is pragmatic in nature, assessing the effect of distal median nerve block, and it is typically performed in CTR without the use of sonography based on our knowledge. The pragmatic research setting can be considered instead as a strength of the research, as it enhances the generalizability of the findings. The results can more readily be applied to real-world treatment practices, thereby increasing their relevance and utility.
While care providers could potentially be blinded to the anesthesia performed, if an external doctor administered it without their knowledge, we did not consider this approach reasoned. This decision was based on the general estimation that the effect of blinding is typically low, 25 and the role of blinding of care providers is likely less significant compared with that of patients.
Patient satisfaction is a crucial factor in modern healthcare. A distal median nerve block may reduce pain during and after CTR, potentially increasing patient satisfaction with the given treatment. It can also be hypothesized that better postoperative pain control may decrease incidence of complications, , such as complex regional pain syndrome, which would be a remarkable achievement. Complex regional pain syndrome, for instance, causes considerable disability, often requiring sick leave and, in some cases, can even lead to a disability pension.. On the contrary, the median nerve can be injured during the injection of the anesthetic agent. Possible advantages or disadvantages of median nerve blockade in addition to local infiltration anesthesia can be assessed by a high-quality randomized controlled clinical trial, which this trial represents.
Most CTRs are performed under local anesthesia, and many surgeons use adjunct median blockade, but there is no evidence behind it. This trial will determine if adjunct median blockade is justified or not. The results of this study can be used to optimize anesthesia for carpal tunnel surgery, improve patient satisfaction, and possibly prevent complications.
Supplemental Material
sj-docx-1-sjs-10.1177_14574969241277028 – Supplemental material for Carpal tunnel release under local anesthesia with or without distal median nerve block: Double-blind randomized clinical trial
Supplemental material, sj-docx-1-sjs-10.1177_14574969241277028 for Carpal tunnel release under local anesthesia with or without distal median nerve block: Double-blind randomized clinical trial by Noora Heikkinen, Yrjänä Nietosvaara, Aleksi Reito, Joonas Sirola, Mikael Hytönen, Aukusti Savolainen and Mikko P. Räisänen in Scandinavian Journal of Surgery
Footnotes
Acknowledgements
We are grateful to research nurse Elina Jalava for her considerable work toward making this trial possible and for the conduction of the trial.
Author contributions
Noora Heikkinen: Planning, writing, and submission of the protocol.
Yrjänä Nietosvaara: Planning, reviewing the protocol, and steering the primary author.
Aleksi Reito: Planning and writing the statistical analysis plan.
Joonas Sirola: Reviewing the protocol and planning the data management and collection.
Mikael Hytönen: Writing the protocol.
Aukusti Savolainen: Writing the protocol.
Mikko P. Räisänen: Planning and writing the protocol. Reviewing the protocol and steering the primary author.
Declaration of conflicting interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Noora Heikkinen, None; Yrjänä Nietosvaara, None; Aleksi Reito, None; Joonas Sirola, None; Mikael Hytönen, None; Aukusti Savolainen, None; Mikko P. Räisänen, Hand and wrist course, Arthrex; and Nerve repair, Axone.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The trial has and will not seek funding from the industry. Investigator Noora Heikkinen received funding from the Vappu Uuspää Foundation in 2021 and 2022–2023 from The Finnish Society for Surgery. The trial sponsors or funders did not have any role in trial design; not in the collection, management, analysis, interpretation of data, writing of the report, or the decision to submit the report for publication.
Ethics approval
The Research Ethics Committee of the Northern Savo Hospital District has approved the trial protocol (2312/2021).
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.