
Abstract
Aims:
Previous studies have reported a ‘smoker’s paradox’, where people who smoke appear to be protected against Severe Acute Respiratory Syndrome CoronaVirus-2 (SARS-CoV-2) infection (COVID-19). This conflicts with well-established evidence that people who smoke are generally more vulnerable to respiratory infections. In this study, we aimed to validate the association between smoking and SARS-CoV-2 infection in a general Dutch population, and to evaluate the evidence underlying the possible causal relationship between smoking and SARS-CoV-2 infection by applying a modern adaptation of the Bradford Hill criteria.
Methods:
In total, 57,833 participants from the Lifelines Cohort Study were included in the analysis. Smoking status, including never smoker, current smoker, and former smoker, was derived from the Lifelines general assessment between 2014 and 2017, while SARS-CoV-2 infection status was derived from an additional COVID-19 questionnaire from 2021 to 2022. Logistic regressions were used for the association between smoking status and infection status. The adapted Bradford Hill’s criteria, including the strength of association (including an analysis of plausible confounding), plausibility, temporality and study design suitability, were applied to evaluate the existing literature.
Results:
We found, compared with never smokers, an increased risk of SARS-CoV-2 infection for former smokers (odds ratio (OR)=1.07, 95% confidence interval (CI)=1.01–1.13), but a reduced risk for current smokers (OR=0.85, 95% CI=0.79–0.92), after adjusting for several relevant covariates. However, we discerned a possible explanation of the smoker’s paradox since we observed that current smokers were more likely to be non-responders to the COVID-19 questions and, more importantly, these non-responders were more likely to have other established risk factors for SARS-CoV-2 infection.
Conclusions:
Introduction
Since the start of the Coronavirus Disease 2019 (COVID-19) pandemic, researchers have published unexpected results on the relationship between smoking and vulnerability to COVID-19; people who smoke appear to be protected against infection with the Severe Acute Respiratory Syndrome CoronaVirus-2 (SARS-CoV-2), the virus that causes COVID-19. This ‘smoker’s paradox’ was, for example, reported in a large meta-analysis showing that current smokers are at reduced risk of SARS-CoV-2 infection with a relative risk (RR) of 0.67 (95% confidence interval (CI) = 0.60–0.75) compared with never smokers [1]. For former smokers, no association was found (RR=0.99, 95% CI=0.94–1.05). Since people who smoke are generally more vulnerable to respiratory infections [2], and smoking has been shown to be associated with poorer COVID-19 outcomes (i.e. hospitalization, mortality) [1,3], this finding led to confusion amongst scientists and healthcare professionals, and received a lot of media attention [4].
To date, the inverse association between smoking and susceptibility to SARS-CoV-2 infection remains disputed. While some studies have raised the possibility of a protective effect from nicotine [3,5 –8], other recent studies showed an increased risk of SARS-CoV-2 infection for people who smoke [9 –11]. This smoker’s paradox may possibly be explained by controversy and serious limitations that exist in the collection and interpretation of all studies. Most importantly, many studies did not adjust for important factors, such as age, gender and socioeconomic position [1]. In addition, most studies were hospital-based, a study design accompanied by various types of bias. It is, for example, possible that patients with severe COVID-19 symptoms stopped smoking due to their sickness or admission to the hospital, and therefore people who smoke may be underrepresented in hospital data [12]. Moreover, some of the heavy smokers may have already died of smoking-related diseases before they reach the age of an average COVID-19 hospital patient (68 years) [13,14].
In this study, first, we aimed to validate the association between smoking and the risk of SARS-CoV-2 infection using a large population-based Dutch cohort. Second, to better evaluate the evidence underlying the possible causal relationship between smoking and SARS-CoV-2 infection, we applied a modern adaptation of the Bradford Hill criteria for causation to the existing literature, including the results from the Dutch population [15,16].
Methods
Lifelines cohort study
To assess the association between smoking and SARS-CoV-2 infection while adjusting for important confounders, we used the Lifelines cohort study [17,18]. The Lifelines study is an ongoing, multidisciplinary, prospective population-based cohort study and biobank, with participants recruited from the three northern provinces of the Netherlands between 2006 and 2013. The Lifelines assessments are conducted every five years, with the most recent assessment (i.e. the second assessment) completed between 2014 and 2017. During the assessment, participants were asked to complete several questionnaires regarding various aspects, including demographics, socioeconomic position, and lifestyle factors, including smoking status. Smoking status was further classified as current smoker, former smoker, or never smoker (description given in the Supplemental material online).
Additionally, questionnaires on COVID-19 were sent on a (bi)weekly basis starting in March 2020 and on a monthly basis starting in July 2020 to the Lifelines adult participants (⩾ 18 years). In total, 26 questionnaires were sent until April 2022. In these questionnaires, participants were repeatedly asked: ‘Have you tested positive for COVID-19?’ and/or ‘What is the result of your COVID-19 test?’. The infection status was defined as ‘infected’ if the participants answered ‘yes’ and/or ‘positive’ to at least one of the questionnaires, and was coded ‘not infected’ when they did not answer ‘yes’ and/or ‘positive’ to any of the questionnaires and answered ‘no’ and/or ‘negative’ to at least one of the questionnaires.
In logistic regression models, we investigated the association between smoking status and risk of SARS-CoV-2 infection adjusted for age, sex, education level, stress level, general health, heavy drinking behaviour and body mass index (BMI). We included these covariates as they have been reported to be the risk factors of SARS-CoV-2 infection and/or COVID-19 progression [19 –23].
Bradford Hill criteria
To assess whether the association between smoking and the risk of SARS-CoV-2 infection is causal, we used a modern adaptation of the Bradford Hill criteria (based on the Shimonovich version [16]). The Bradford Hill criteria include nine criteria by which to establish epidemiological evidence of a causal relationship (strength, consistency, specificity, temporality, biological gradient, plausibility, coherence, experiment and analogy). Investigating causality is important, since statistical associations do not necessarily imply causation. For example, Bradford Hill argued that the presence of biological plausibility can increase the likelihood of causation.
The Bradford Hill criteria have been widely used in public health research [15,16], and there are several modern adaptations of Bradford Hill’s criteria. In general, they agree that to assess causality, the following criteria should be evaluated: the strength of association (including an analysis of plausible confounding), plausibility, temporality and study design suitability [16]. We reviewed the existing literature, including our Lifelines study, based on these four criteria. The existing literature commenced with the large meta-analysis of Simons et al. [1] as a basis, which examined the association between smoking status and risk of SARS-CoV-2 infection. In addition, we added relevant studies published after the publication date of this meta-analysis.
Results
A total of 57,833 Lifelines participants with valid data on smoking status were included in this study, of whom 10,510 reported to have been infected with the SARS-CoV-2 virus, 22,678 reported to not have been infected and 24,645 did not report their infection status (Supplemental Figure 1 and Supplemental Table I). When we excluded the participants who did not report their infection status, the fully adjusted logistic regression analysis showed that former smokers had an increased risk of SARS-CoV-2 infection compared with never smokers (odds ratio (OR)=1.07, 95% CI=1.01–1.13). In contrast, current smokers were less likely to be infected than never smokers (OR=0.85, 95% CI=0.79–0.92) (Supplemental Table II). A sensitivity analysis with an extra category including those who did not report the infection status showed that current smokers were more likely to not report their infection status (OR=1.10, 95% CI=1.03–1.16) compared with never smokers (Supplemental Tables III and IV). Moreover, among the non-responders, the current smokers were more often male, lower educated, heavy drinker and reported poorer health conditions than the never smokers (Supplemental Table V).
Discussion
Despite the emerging discussion on the smoker’s paradox that current smoking is inversely associated with SARS-CoV-2 infection, this study strived to provide new insights into this paradox.
Although our original empirical evidence also revealed an inverse association between smoking and SARS-CoV-2 infection, we showed that selection bias could be the underlying reason for the existence of the paradox as current smokers were more likely to not respond to the COVID-19 questions. Moreover, people who smoke and did not respond to the COVID-19 questions were more likely to have other established risk factors for SARS-CoV-2 infection, such as male gender, lower education and heavy drinker. This is important since these factors were previously reported as risk factors for SARS-CoV-2 infection [21 –23]. Although we adjusted for these factors in our analysis, residual confounding may still exist when the selection effect is strong. This selection effect could therefore be a possible explanation for the reduced risk of infection found in current smokers.
The main strength of this piece of empirical evidence using Lifelines was the use of a large population-based cohort, including information on important confounders. As investigating the association between smoking and SARS-CoV-2 infection is challenging, we also encountered some limitations using the Lifelines data. First, we observed an underrepresentation of current smokers in our study compared with the general Dutch population (14% versus 21% [24]). In addition, the smoking status was derived from the second general assessment from 2014 to 2017, which might have changed in the intermediate years until the assessment of COVID-19 status in 2021 and 2022. Last, the outcome variable of the Lifelines study is self-reported diagnosed SARS CoV-2-infection, which differs from SARS CoV-2-infection as there might be undiagnosed SARS CoV-2-infection and other COVID-19 related health outcomes, such as severe COVID-19 and COVID-19 hospitalization. It is crucial to acknowledge that these different outcomes may have different profiles of risk factors.
Bradford Hill criteria
To further scrutinize the association between smoking and the risk of SARS-CoV-2 infection integrating evidence from this study with previous published literature, we reviewed the existing literature using a modern adaptation of the Bradford Hill criteria for causation. We analysed four aspects, including the strength of association (confounding), plausibility, temporality and study design suitability.
Strength and consistency
The strength of the association between smoking and SARS-CoV-2 infection has been examined in various case–control studies, cohort studies and a meta-analysis in a variety of populations. Table I shows an overview of these studies, including their main results [1,9,11,25]. There was high between-study heterogeneity and limited consistency in the study results, probably due to variation in methodological rigour, variability in sample sizes and variation in determination of smoking status and COVID-19 outcomes.
Strength of the association between smoking and risk of SARS-CoV-2 infection in the current study and other published studies a .
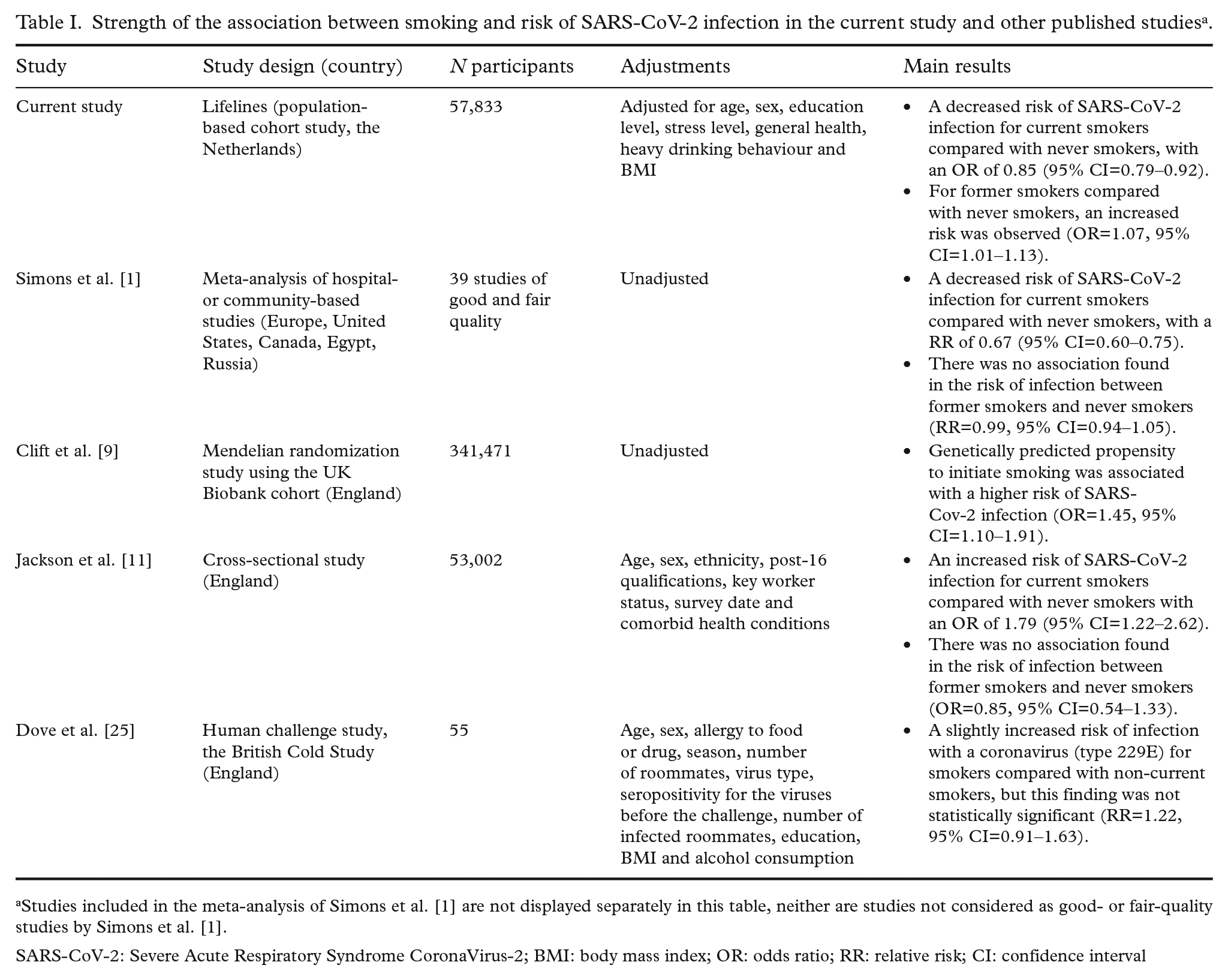
Studies included in the meta-analysis of Simons et al. [1] are not displayed separately in this table, neither are studies not considered as good- or fair-quality studies by Simons et al. [1].
SARS-CoV-2: Severe Acute Respiratory Syndrome CoronaVirus-2; BMI: body mass index; OR: odds ratio; RR: relative risk; CI: confidence interval
Confounding
In our Lifelines study, the baseline analysis was adjusted for age and sex, and we used different models to assess the influence of several covariates (education, stress score, self-reported general health status, heavy drinking behaviour and BMI) on the association between smoking and risk of SARS-CoV-2 infection (Supplemental Table II). We did not observe significant differences between the models, indicating no meaningful confounding of these covariates. Jackson et al. [11] did observe moderation by socioeconomic position, showing a higher risk of infection for current smokers compared with never smokers among people with a lower education, but no differences were observed among people with a higher education.
The possibility for residual mediators, modifiers and confounders exists. For example, we and others did not have information on the number of polymerase chain reaction (PCR) tests taken. If the number of tests is related to smoking status or other (non-measured) confounding factors, this could influence the results. This is not unlikely, for example, since people who smoke more often have a cough, they may test more often. Or the other way around, it is also possible that people who smoke may test less often as they could believe their cough is caused solely by smoking.
A study design that provides better controls for residual confounding is Mendelian randomization, where genotype information is used as an instrumental variable. Genome-wide association studies have identified genetic risk variants that are associated with smoking initiation and/or smoking heaviness [26]. Because genetic risk variants are randomly assigned during meiosis, they should not be associated with confounders and can be used as a ‘proxy’ for smoking. So far, one Mendelian randomization study has been performed to investigate the association between smoking and the risk of SARS-CoV-2 infection, showing higher risks for current smokers [9].
Plausibility
The biological plausibility of the association between smoking and SARS-CoV-2 infection remains unclear. Some hypotheses have proposed a harmful effect of smoking, while others claim a protective effect of nicotine on the risk of SARS-CoV-2 infection.
Mechanisms supporting an increased vulnerability for people who smoke
There are multiple biological mechanisms known through which smoking may increase the risk of infection with the SARS-CoV-2 virus. First, it is well-known that smoking increases the risk of respiratory infections, both viral and bacterial [2]. Cigarette smoke lowers the immunity of the respiratory system by causing inflammation and peribronchial fibrosis, impaired mucociliary clearance, and disruption of the respiratory epithelium [2,27,28]. Smoking also leads to a decreased function of neutrophils, alveolar macrophages and T-cells, resulting in an impaired host immunity [27,29].
Besides biological mechanisms, behavioural factors involved in smoking may increase the risk of SARS-CoV-2 infection and transmission in current smokers. For example, smoking involves regular hand-to-mouth movements and tobacco products are sometimes shared between individuals, facilitating viral transmission [7,30].
Mechanisms supporting a decreased vulnerability for people who smoke
A controversial theory suggests that nicotine might be able to prevent infection with the SARS-CoV-2 virus due to its anti-inflammatory effects [5]. Nicotine inhibits the production of pro-inflammatory cytokines such as TNF, IL-1 and IL-6, without inhibition of anti-inflammatory cytokines such as IL-10. Continuous suppression of these systemic cytokines in smokers may adapt their immune response to becoming more tolerant and less reactive to continuous inflammatory stimuli [5 –7]. Another theory suggests that the nicotinic acetylcholine receptors, targets of nicotine, contribute to SARS-CoV-2 infection by acting as SARS-CoV-2 Spike co-receptors [8]. Nicotine might compete with the virus for surface binding and thereby reduce SARS-CoV-2 entry [3]. However, to date, any protective role for nicotine is still speculative.
The role of the ACE-II receptor
It is known that the SARS-CoV-2 virus enters the epithelial cells through binding to the angiotensin converting enzyme 2 (ACE-II) receptor, a receptor expressed on several human tissues, including lung tissue [31]. It has been proposed that smoking modulates expression of the ACE-II receptor, but its precise effect remains unclear. Some studies showed that people who smoke have increased expression of ACE-II [32 –36] (which could relate to an increased risk of infection), while others showed that smoking and nicotine contribute to downregulation of ACE-II [37]. A study from Porter et al. [38] found that cigarette smoke induces ACE-II expression, but does not alter the efficiency of cellular SARS-CoV-2 infection, suggesting that current smokers do not have an increased risk of infection. As ACE-II may play an important role in vulnerability to COVID-19 in people who smoke, further research is warranted to explore this potential mechanism.
Temporality and study design suitability
Temporality refers to the necessity that the effect must occur after the cause. As previously mentioned, most studies assessing the association between smoking and the risk of SARS-CoV-2 infection were cross-sectional. Cross-sectional studies are known to be susceptible to reverse causation, in which the outcome precedes and causes the exposure. In the case of COVID-19, it is possible that patients with COVID-19 alter their smoking behaviour because of their sickness or due to their admission to hospital [12]. A way to be more certain about the temporal sequence is to conduct prospective studies or Mendelian randomization studies, in which the samples are studied prior to the onset of the disease and followed over time. With the current study, we were one of the first assessing the association between smoking and the risk of SARS-CoV-2 infection using a prospective design, but there is a need for better designed prospective cohort studies with least methodological limitations possible to better prepare for future pandemics, such as Mendelian randomization studies with randomized sampling of the general population.
Conclusion
We argue that there is insufficient evidence for an inverse association between smoking and the risk of SARS-CoV-2 infection, as we observed high inconsistency between study results, a high possibility for residual confounding, and no clear evidence for biological plausibility. Furthermore, assessing this association is challenging due to epidemiological issues arising in this subject, such as misclassification of people who smoke due to self-report, residual bias and incompleteness of COVID-19 data.
To accurately assess the association between smoking status and the risk of SARS-CoV-2 infection, there is a need for data from high-quality studies. These studies should include linkage with the PCR-confirmed testing results from national healthcare registries to avoid self-reporting bias and require a comprehensive collection of covariates that could interfere with the association between smoking and infection (e.g. socioeconomic position, number of COVID-19 tests). Finally, the mechanisms underlying the susceptibility of smokers to SARS-CoV-2 infection requires further exploration, including the potential roles of nicotine and ACE-II.
Supplemental Material
sj-docx-1-sjp-10.1177_14034948241253690 – Supplemental material for New insights into the paradox between smoking and the risk of SARS-CoV-2 infection (COVID-19): Insufficient evidence for a causal association
Supplemental material, sj-docx-1-sjp-10.1177_14034948241253690 for New insights into the paradox between smoking and the risk of SARS-CoV-2 infection (COVID-19): Insufficient evidence for a causal association by Iris Kramer, Yinjie Zhu, Naomi A. Van Westen-Lagerweij, Louise H. Dekker, Jochen O. Mierau and Esther A. Croes in Scandinavian Journal of Public Health
Footnotes
Acknowledgements
Lifelines Corona Research initiative: H. Marike Boezen† (Department of Epidemiology, University of Groningen, University Medical Centre Groningen, Groningen, The Netherlands), Jochen O. Mierau (Department of Economics, Econometrics & Finance, Faculty of Economics and Business, University of Groningen, Groningen, The Netherlands; Lifelines Cohort Study and Biobank, Groningen, The Netherlands; Team Strategy & External Relations, University of Groningen, University Medical Centre Groningen, The Netherlands), H. Lude Franke (Department of Genetics, University of Groningen, University Medical Centre Groningen, Groningen, The Netherlands), Jackie Dekens (Department of Genetics, University of Groningen, University Medical Centre Groningen, Groningen, The Netherlands; Centre of Development and Innovation, University of Groningen, University Medical Centre Groningen, Groningen, The Netherlands), Patrick Deelen, Pauline Lanting (Department of Genetics, University of Groningen, University Medical Centre Groningen, Groningen, The Netherlands), Judith M. Vonk, Ilja Nolte (Department of Epidemiology, University of Groningen, University Medical Centre Groningen, Groningen, The Netherlands), Anil P.S. Ori (Department of Genetics, University of Groningen, University Medical Centre Groningen, Groningen, The Netherlands; Department of Psychiatry, University of Groningen, University Medical Centre Groningen, Groningen, The Netherlands), Annique Claringbould, Floranne Boulogne, Marjolein X.L. Dijkema, Henry H. Wiersma, Robert Warmerdam, Soesma A. Jankipersadsing (Department of Genetics, University of Groningen, University Medical Centre Groningen, Groningen, The Netherlands), Irene van Blokland (Department of Genetics, University of Groningen, University Medical Centre Groningen, Groningen, The Netherlands; Department of Cardiology, University of Groningen, University Medical Centre Groningen, Groningen, The Netherlands), Geertruida H. de Bock (Department of Epidemiology, University of Groningen, University Medical Centre Groningen, Groningen, The Netherlands), Judith GM Rosmalen (Department of Psychiatry, University of Groningen, University Medical Centre Groningen, Groningen, The Netherlands; Department of Internal Medicine, University of Groningen, University Medical Centre Groningen, Groningen, The Netherlands), Cisca Wijmenga (Department of Genetics, University of Groningen, University Medical Centre Groningen, Groningen, The Netherlands).
† = deceased.
Author contributions
Iris Kramer: conceptualization, methodology, writing – original draft, project administration. Yinjie Zhu: conceptualization, methodology, formal analysis, writing – review and editing. Naomi A. van Westen-Lagerweij: conceptualization, methodology, writing – review and editing. Louise H. Dekker: conceptualization, methodology, writing – review and editing, supervision. Jochen O. Mierau: conceptualization, writing – review and editing, supervision. Esther A. Croes: conceptualization, writing – review and editing, supervision, funding acquisition. Lifelines Corona Research Initiative: resources, investigation.
Consent to participate
Informed consent was obtained from all individual participants included in the study.
Declaration of conflicting interests
The authors have no conflicts of interest to declare.
Ethics approval
The Lifelines study was conducted according to the principles of the Declaration of Helsinki and approved by the Medical Ethics Committee of the Institutional Review Board of the University Medical Centre Groningen, The Netherlands (2007/152).
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Dutch Ministry of Health, Welfare and Sport (grant number: 41-8007 – WP 34). The Lifelines initiative has been made possible by subsidy from the Dutch Ministry of Health, Welfare and Sport, the Dutch Ministry of Economic Affairs, the University Medical Centre Groningen (UMCG), Groningen University and the Provinces in the north of the Netherlands (Drenthe, Friesland, Groningen).
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.