
Abstract
Aquaporin-4 (AQP4)-mediated water transport at astrocytic end-feet is pivotal for glymphatic clearance, a process increasingly recognized as a determinant of brain health and resilience to neurodegeneration. Nevertheless, existing literature has not yet systematically clarified how sex hormones influence AQP4 biology and, in turn, glymphatic efficiency, leaving a critical gap in our understanding of sex-specific vulnerability to disorders such as Alzheimer's disease. To address this gap, we investigated how gonadal hormones influence AQP4 expression and polarity within the context of neuroinflammatory processes, drawing on evidence from preclinical models. We conducted a comprehensive review of in vivo and in vitro studies across ischemic stroke, traumatic brain injury, hypoxia-ischemia, osmotic stress, and viral neuroinflammation models, extracting standardized data on hormonal status, AQP4 metrics, neuroinflammatory markers, and fluid-clearance outcomes. The collated findings reveal that loss of estrogen, progesterone, or testosterone amplifies microgliosis, NF-κB activation, cytokine release (IFN-γ, IL-6, IL-8), and AQP4 mislocalization, whereas physiological hormone replacement reverses these changes, restores AQP4 polarity, and stabilizes the blood-brain barrier. These results indicate that sex-dependent regulation of AQP4 and glymphatic flow is a plausible contributor to the higher incidence and faster progression of Alzheimer's disease in postmenopausal women. Our synthesis underscores the need for real-time glymphatic imaging combined with targeted hormonal or anti-inflammatory interventions to determine whether re-establishing proper hormone signaling or AQP4 polarity can slow proteopathic accumulation and modify disease trajectories.
Introduction
The glymphatic system is essential in maintaining brain homeostasis by facilitating the transport of cerebrospinal fluid (CSF) and clearing interstitial solutes, including potentially neurotoxic proteins such as amyloid-β. 1 Glymphatic function critically depends on aquaporin-4 (AQP4), the predominant water channel in the central nervous system (CNS). 2 AQP4 is highly concentrated in astrocyte endfeet surround cerebral microvessels, creating a vital site for fluid exchange. 3 Disruptions to AQP4 expression or localization can have profound effects on cerebrovascular integrity, edema formation, and overall neurological outcome. 4
Research on AQP4 has long highlighted its dual role in fluid regulation. When properly controlled, it supports fluid homeostasis and promotes the resolution of edema. 5 However when its function is disrupted, it can worsen both cytotoxic and vasogenic edema. 6 Importantly, multiple lines of evidence point to sex hormones as key modulators of AQP4. Studies of ischemic stroke consistently show that female rodents, or males receiving exogenous female sex hormones, often exhibit smaller infarct volumes, reduced edema, and improved neurological outcomes.7,8 Conversely, the influence of testosterone on AQP4 is comparatively less explored but has been shown to upregulate this channel in cultured astrocytes. 9
The reason for focusing on these hormone-AQP4 interactions in the context of the glymphatic system arises partly from clinical observations in Alzheimer's disease. This neurodegenerative disorder is notably more prevalent in women than in men. 10 While numerous factors likely contribute to this sex difference, the glymphatic hypothesis posits that impaired clearance of neurotoxic proteins via AQP4 channels could accelerate disease progression. Whether and how sex hormones shape glymphatic function through regulation of AQP4, and whether this might relate to Alzheimer's disease risk, remains an open question. This review aims to synthesize the extensive experimental data on estrogen, progesterone, and testosterone in modulating AQP4 under pathological conditions, and then frame these findings within the broader discussion of glymphatic activity and Alzheimer's disease prevalence in women.
Hormonal regulation of aquaporin-4: implications for glymphatic function
In principle, glymphatic efficiency requires a coordinated process of arterial pulsation, intact perivascular spaces, and functional astrocyte endfeet equipped with properly regulated AQP4. 11 When pathological insults such as stroke or TBI, overwhelm these regulatory mechanisms, water flux can become detrimental if it favors excessive cellular swelling and edema.12,13 Understanding how each hormone modifies AQP4 expression offers clues as to how male and female brains might differentially handle fluid shifts in both health and disease.
The collective evidence from in vitro and in vivo models suggests that estrogen and progesterone generally reduce harmful edema by modulating AQP4 expression and preserving blood-brain barrier (BBB) integrity,7,8,12,14,15 whereas testosterone has been found to upregulate AQP4 in cultured astrocytes under certain conditions. 9 It thus appears that female hormones often protect the brain from maladaptive AQP4 changes, an effect that could foster more stable glymphatic function during or after injury. By contrast, testosterone's net impact on glymphatic clearance remains less clearly defined. Its enhancement of AQP4 might favor improved fluid outflow or, conversely, could lead to harmful astrocyte swelling if BBB disruption coincides with excessive channel expression. These differential hormonal influences on AQP4 expression and their potential implications for glymphatic function will be explored in greater detail in the subsequent sections of this review. Table 1 summarizes key experimental studies investigating the effects of sex hormones on AQP4 expression and brain edema under various pathological conditions, highlighting hormone-specific outcomes across both in vitro and in vivo models.
Summary of experimental studies examining the effects of sex hormones on AQP4 expression and brain edema under pathological conditions.
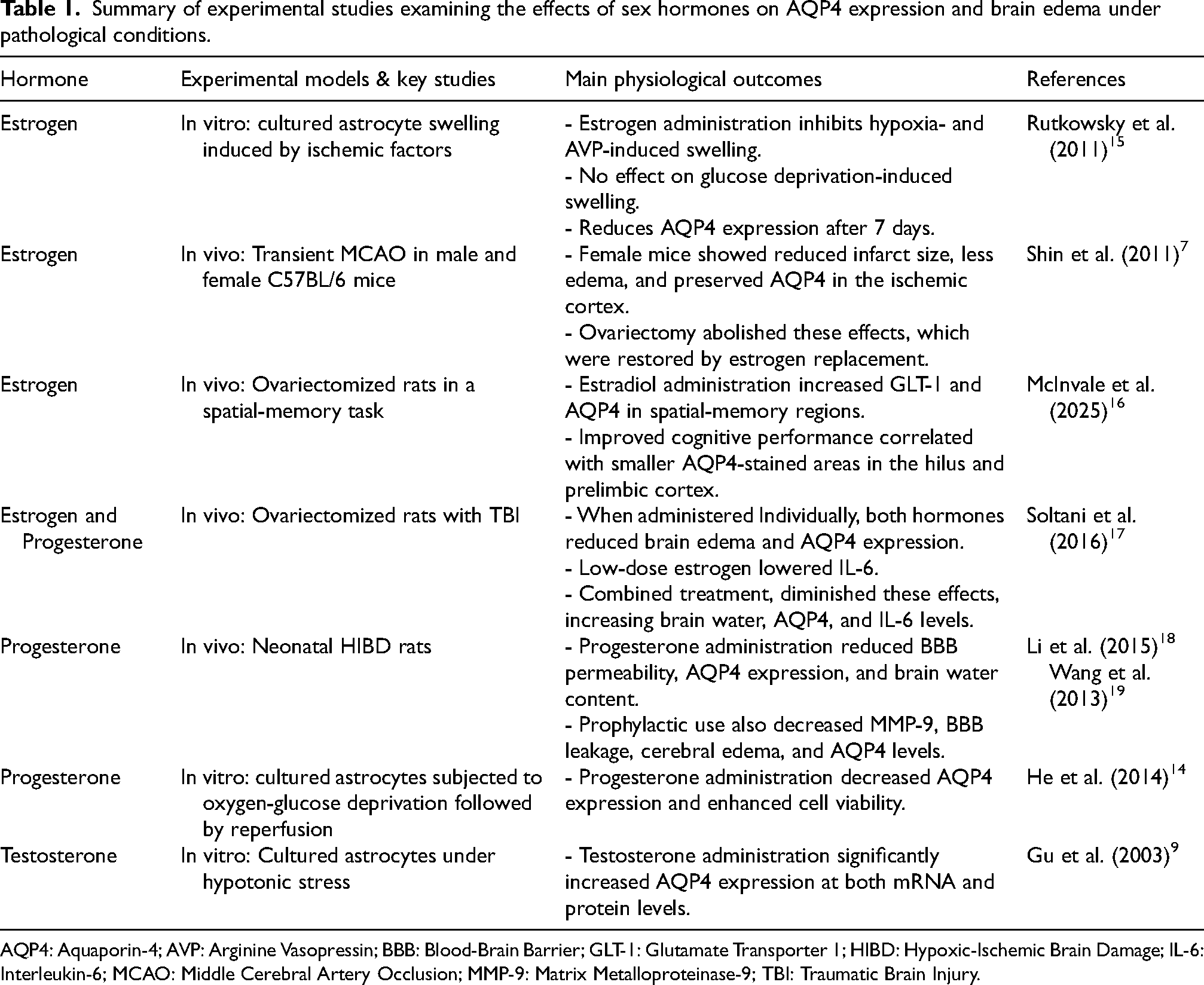
AQP4: Aquaporin-4; AVP: Arginine Vasopressin; BBB: Blood-Brain Barrier; GLT-1: Glutamate Transporter 1; HIBD: Hypoxic-Ischemic Brain Damage; IL-6: Interleukin-6; MCAO: Middle Cerebral Artery Occlusion; MMP-9: Matrix Metalloproteinase-9; TBI: Traumatic Brain Injury.
Estrogen and AQP4
Numerous experimental models spotlight estradiol as a potent protector against cerebral edema. Jennifer M. Rutkowsky et al. examined the effect of estradiol in vitro on astrocyte swelling induced by ischemic factors such as hypoxia, aglycemia, and arginine vasopressin (AVP). Estradiol abolished the swelling triggered by hypoxia or AVP, though not by glucose deprivation, and reduced AQP4 protein abundance after seven days of exposure. These findings emphasize the importance of timing, as short-term estradiol treatments did not consistently reduce AQP4 expression, while longer interventions resulted in a clear decline in AQP4 protein levels. 15
In a separate model of transient middle cerebral artery occlusion (MCAO) comparing male and female C57BL/6 mice, females exhibited smaller infarct volumes, less edema, and a relative preservation of AQP4 expression in the ischemic cortex. When female mice were ovariectomized, these protective effects disappeared, and they were restored, however, by estrogen replacement therapy. Moreover, using an estrogen receptor (ER) antagonist reversed the previously observed protective benefits, implicating estrogen receptor-mediated signaling in stabilizing AQP4 levels. 7 In a related study, ER-β activation was shown to attenuate BBB disruption, in part through suppression of the HIF-1α/VEGF signaling pathway. 20 This evidence may seem contradictory, with one study reporting that estradiol reduces AQP4 levels, while another finds that it maintains AQP4 expression. The reason for this could be that estradiol helps avert injurious patterns of AQP4 dysregulation, either limiting its pathological upregulation or preventing its detrimental loss, depending on the specific injury context and phase of edema progression.
Nonetheless, a recent behavioral study offers an important insight. McInvale J.J. et al. showed that ovariectomized rats given 17β-estradiol (4.5 or 45 µg kg−1) 24–48 h before a delayed-spontaneous-alternation task showed enhanced spatial working memory and region-specific shifts in astrocytic markers. Estradiol increased GLT-1 and AQP4 in spatial-memory regions (prelimbic cortex and dorsal hippocampus) relative to the dorsolateral striatum, yet within the estradiol group, better task performance correlated with smaller AQP4-stained area in the hilus and prelimbic cortex. The authors interpret this negative correlation as evidence of greater AQP4 polarization to perivascular end-feet, an arrangement that supports efficient water flux despite an overall reduction in diffuse staining. Thus, estradiol appears to decrease bulk AQP4 signal while sharpening its localization, aligning with the concept that polarity, not quantity per se, predicts cognitive benefit. This also supports the idea that estradiol refines AQP4 distribution in a region-specific manner. 16
Although these studies do not directly measure glymphatic flux, it is reasonable to suggest that estrogen's regulation of AQP4 promotes more physiological water transport through astrocyte endfeet, thereby creating a supportive environment for effective solute clearance. In humans, the higher incidence of Alzheimer's disease among postmenopausal women may partly derive from the sudden drop in circulating estradiol and the consequent loss of its protective effects on fluid homeostasis. While this theory remains to be tested directly in glymphatic research, the preclinical data broadly suggest that estrogen fosters a more balanced and beneficial AQP4 profile.
Progesterone and AQP4
Progesterone's capacity to reduce cerebral edema has also been demonstrated across several models of brain injury. In a study conducted by Z. Soltani and colleagues using ovariectomized female Wistar rats with experimentally induced traumatic brain injury (TBI), researchers assessed how estrogen and progesterone, given alone or in combination, affected brain water content, AQP4 expression, and interleukin-6 (IL-6) levels. When administered individually, both hormones lowered brain edema and reduced AQP4 expression, and low-dose estrogen also reduced IL-6 levels. However, combining estrogen and progesterone partially canceled out these protective effects, leading to higher brain water content and increased AQP4 and IL-6 compared to single-hormone treatment. Overall, the authors concluded that either hormone alone appears more effective at alleviating brain edema than combining the two, potentially through modulation of AQP4 and IL-6. 17
Moreover, in 7-day-old neonatal Wistar rats with hypoxic-ischemic brain damage (HIBD), progesterone again significantly lowered BBB permeability, decreased AQP4 expression, and reduced brain water content relative to untreated controls. The BBB and AQP4 levels rose early in the injury process within six hours, peaked around 72 h, and slowly returned to normal. Progesterone effectively alleviated the aberrant surge in AQP4, underscoring the hormone's role in preventing maladaptive astrocyte swelling. 18 A related investigation in 7-day-old Wistar rats with HIBD further demonstrated that prophylactic progesterone administration reduces MMP-9 generation, BBB leakage, cerebral edema, and AQP4 expression. These neuroprotective effects appear to be mediated by the downregulation of both AQP4 and MMP-9 in the cerebral cortex, underscoring progesterone's critical role in maintaining BBB integrity and mitigating edema following hypoxic-ischemic injury. 19 This study suggests that by limiting MMP-9 upregulation, progesterone helps maintain BBB integrity, which indirectly reduces the fluid load on astrocytes. As a result, astrocytes may not need to increase AQP4 expression to manage excessive water influx.
Additionally, certain mechanistic studies in cultured astrocytes highlight protein kinase C (PKC) signaling as a key mediator of progesterone's effect on AQP4. When astrocyte cultures were subjected to oxygen-glucose deprivation followed by reperfusion, a common in vitro model for ischemia/reperfusion injury, progesterone reduced AQP4 expression and improved cell viability. However, pharmacologic inhibition of PKC negated progesterone's downregulatory effect on AQP4, pointing to a direct PKC-linked pathway. 14
From a glymphatic standpoint, progesterone might be expected to facilitate normal fluid exchange when the BBB is intact and to prevent the pathological changes that drive edema. It remains largely speculative whether these mechanisms lead to enhanced glymphatic clearance or contribute to sex-based differences in neurodegenerative disease risk. However, they support the notion that female sex hormones may play a long-term role in maintaining physiological water balance in the brain.
Testosterone and AQP4
Compared to estrogen and progesterone, testosterone's role in AQP4 dynamics is less extensively reported. One study by Gu et al. focusing on cultured astrocytes found that testosterone significantly upregulated AQP4 at both mRNA and protein levels. 9 This upregulation appeared protective under hypotonic stress, as astrocytes were more resilient to osmotic swelling when AQP4 expression was heightened. Interestingly, the addition of a protein kinase C activator, which can quickly reduce AQP4 transcription, 21 diminished the upregulatory effect of testosterone, 9 emphasizing that the hormone's influence on AQP4 is tied to specific intracellular signaling cascades.
Whereas estrogen and progesterone often mitigate pathological processes, testosterone's effect could be more context-dependent. During the early phases of injury, if BBB breakdown occurs, an elevated AQP4 expression might actually favor astrocyte swelling. Conversely, in a later recovery or maintenance phase, enhanced AQP4-mediated fluid clearance could help remove excess interstitial fluid. Thus, whether testosterone confers a net protective or detrimental effect might hinge on injury timing, hormone levels, and the overall state of the neurovascular unit. The regulatory effects of sex hormones on AQP4 expression and polarization are summarized in Figure 1, which also depicts the putative intracellular signaling mechanisms associated with each hormone.
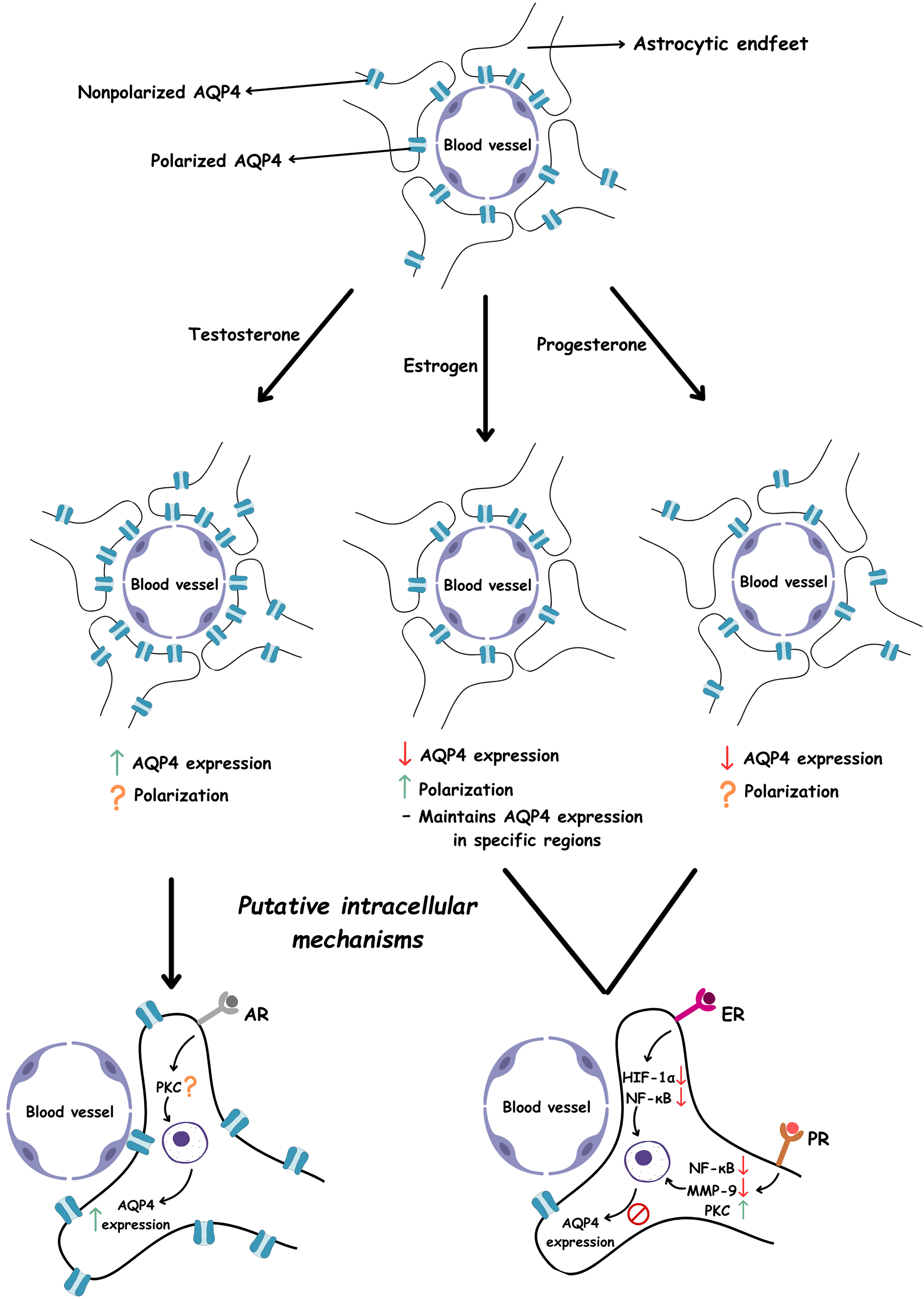
Illustration of the effects of testosterone, estrogen, and progesterone on AQP4 expression and polarization, along with their putative intracellular mechanisms. Estrogen reduces the expression of HIF-1α and NF-κB, both of which are known to upregulate AQP4. This downregulation ultimately leads to decreased AQP4 expression. Progesterone suppresses NF-κB and MMP-9, factors associated with increased AQP4 levels, and concurrently activates the PKC pathway, a known negative regulator of AQP4. These combined actions contribute to the downregulation of AQP4. In contrast, testosterone has been shown to upregulate AQP4 expression. However, its precise intracellular mechanism remains unclear. Notably, evidence suggests that the PKC pathway may be involved in testosterone's effect on AQP4. ↑ indicates increase, ↓ indicates decrease, ? indicates unknown.
Potential links to glymphatic function and Alzheimer's disease
A key motivation for examining these hormone-AQP4 interactions is the observed higher prevalence of Alzheimer's disease in women, a phenomenon often attributed to multiple influences 10 such as longer average lifespan, genetic factors, and postmenopausal decline in estrogen and progesterone. Given that the glymphatic system is responsible for clearing proteins, including amyloid-beta, any sustained dysregulation of AQP4 in postmenopausal women might reduce clearance capacity and accelerate pathological accumulation.
The studies summarized here, while primarily involving acute injury models, collectively reinforce the principle that estrogen and progesterone help maintain healthy AQP4 functioning in astrocytes, protect against severe fluid imbalances, and stabilize the BBB. Even in the absence of overt injury, it is plausible that these hormones could help regulate normal glymphatic flow. For this reason, the postmenopausal decline in estrogen and progesterone concerning AQP4 functioning could be proposed as one reason why Alzheimer's disease occurs more frequently in older women than in men. However, this explanation may not apply to young-onset Alzheimer's disease (YOAD), which begins before age 65 and represents approximately 5% of all Alzheimer's disease diagnoses, 22 because these hormones remain relatively higher in premenopausal women. As supporting evidence, a recent systematic review assessing whether women have a higher prevalence or incidence of YOAD than men found no statistically significant sex differences. 22
On the other hand, the exact impact of testosterone on chronic glymphatic activity remains uncharacterized, although its capacity to increase AQP4 in vitro suggests a possible role in augmenting fluid exchange under some conditions. 9
It is important to emphasize that no direct measurements of glymphatic flux (such as in vivo tracer clearance assays) were performed in these cited studies. Nevertheless, an expanding range of techniques now enables quantitative or semi-quantitative evaluation of glymphatic transport in both animal models and human subjects,23–27 as summarized in Figure 2. As a result, it remains speculative to conclude that glymphatic function is definitively more robust in one sex than the other based solely on hormone-mediated changes in AQP4. In the future, combining these imaging modalities with artificial intelligence and machine learning holds great promise for improving flux quantification, enabling automated pattern recognition, and advancing the identification of sex-specific factors influencing glymphatic function. Still, the consistent theme of female sex hormones promoting more favorable AQP4 regulation supports the broader hypothesis that premenopausal women might benefit from superior fluid and waste clearance over the long term, whereas postmenopausal women lack a parallel level of hormonal protection. To better understand the role of sex hormones in the glymphatic system, it is also important to investigate their influence on CNS immunity and neuroinflammatory processes.
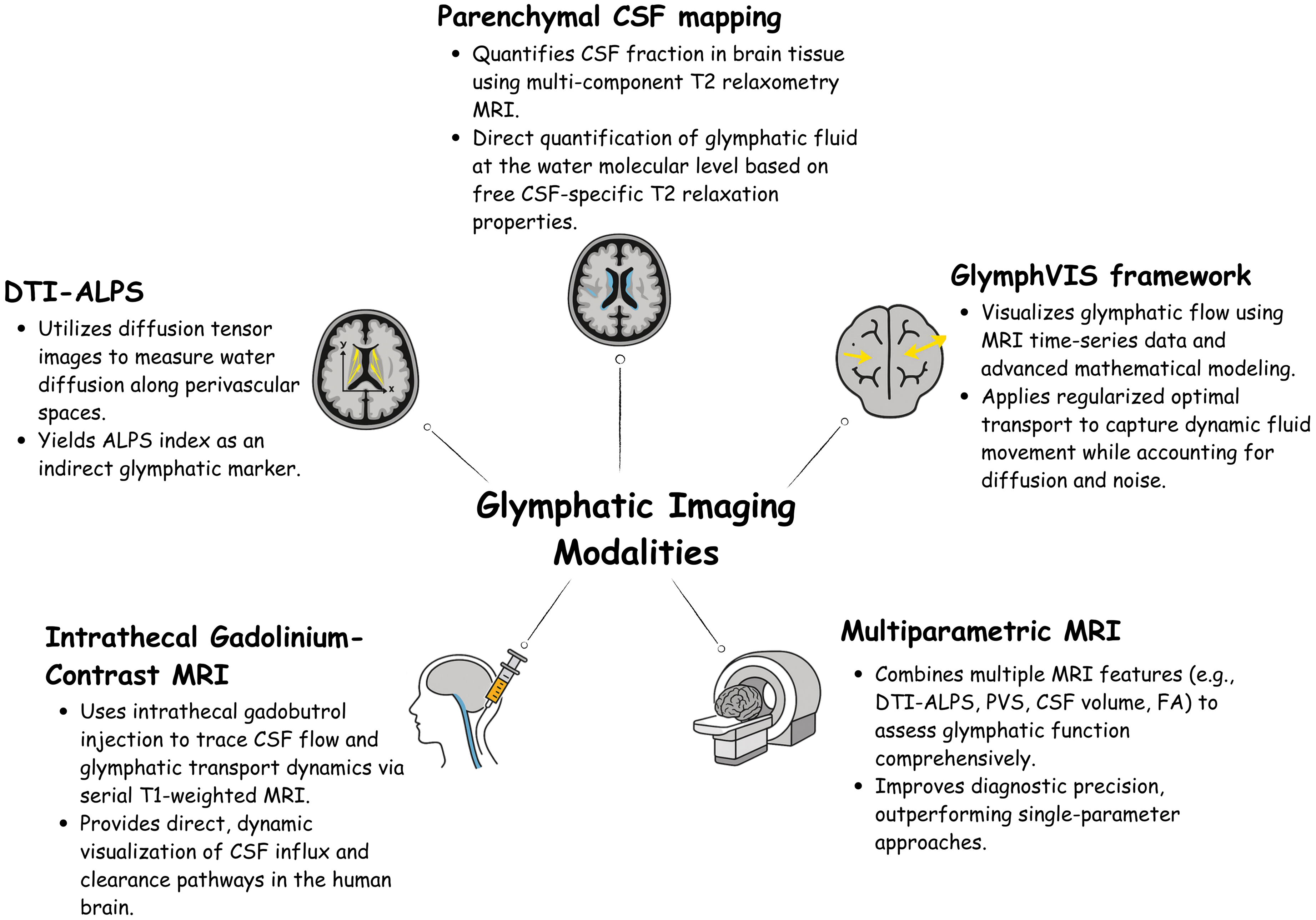
Modern methods for visualizing and analyzing the structure and function of the glymphatic system.
Sex hormones, AQP4, and neuroinflammation
A growing body of work shows that the principal gonadal hormones such as estrogens, progesterone, and testosterone, are pivotal regulators of the immunity within the human body. 28 Acting through classic nuclear and membrane receptors, these hormones restrain NF-κB signaling, mitigate microglial activation, and limit oxidative stress. 28 Estradiol, by engaging ER-α/β, suppresses pro-inflammatory gene transcription and up-regulates astrocytic glutamate transporters. 29 For instance, estrogen binds to and activates the ERα receptor, which triggers cytoplasmic signaling pathways such as PI3 K activation. This estrogen-ERα activity inhibits the inflammatory activation of NF-κB, preventing its translocation into the nucleus. As a result, the production of proinflammatory mediators is reduced. However, in a low-estrogen environment, ERα is not activated, and this anti-inflammatory effect is lost (Figure 3). Consequently, when cytokine ligands bind to their receptors (such as TNFR or IL-1R), they initiate the canonical NF-κB pathway. 30 This pathway begins with the activation of the IKK complex, which consists of two catalytic subunits (IKKα and IKKβ) and a regulatory subunit (NEMO/IKKγ). The IKK complex becomes activated by various stimuli, including cytokines, microbial components, growth factors, mitogens, and stress signals. Once activated, IKK phosphorylates IκB-α at two specific serine residues, marking it for ubiquitin-dependent degradation by the proteasome. This degradation releases NF-κB dimers, primarily p50 and p65, allowing them to translocate into the nucleus and activate the transcription of inflammatory genes. 31 Ovariectomy studies support this view by showing that estrogen withdrawal heightens microglial activation, Aβ deposition, and NF-κB translocation, whereas physiological E2 replacement reverses these changes in models of Alzheimer's disease and systemic LPS stimuli. 30 Likewise, progesterone delivered after repetitive mild traumatic brain injury achieves a similar neuroprotective profile, stabilizing the BBB, inhibiting NF-κB signaling, and preventing MMP-driven AQP4 surges that foster edema. 32 Analogously, it was shown that testosterone replacement in hypogonadal men lowers TNF-α, IL-1β, and CRP and can elevate the anti-inflammatory cytokine IL-10. 33 Experimental data extend these observations to viral neuroinflammation. Zika virus infection in male mice reduces circulating testosterone and precipitates brain inflammation, yet post-infection testosterone supplementation improves survival, lessens neural injury, and diminishes CD8+ T-cell infiltration and IFN-γ production. 34
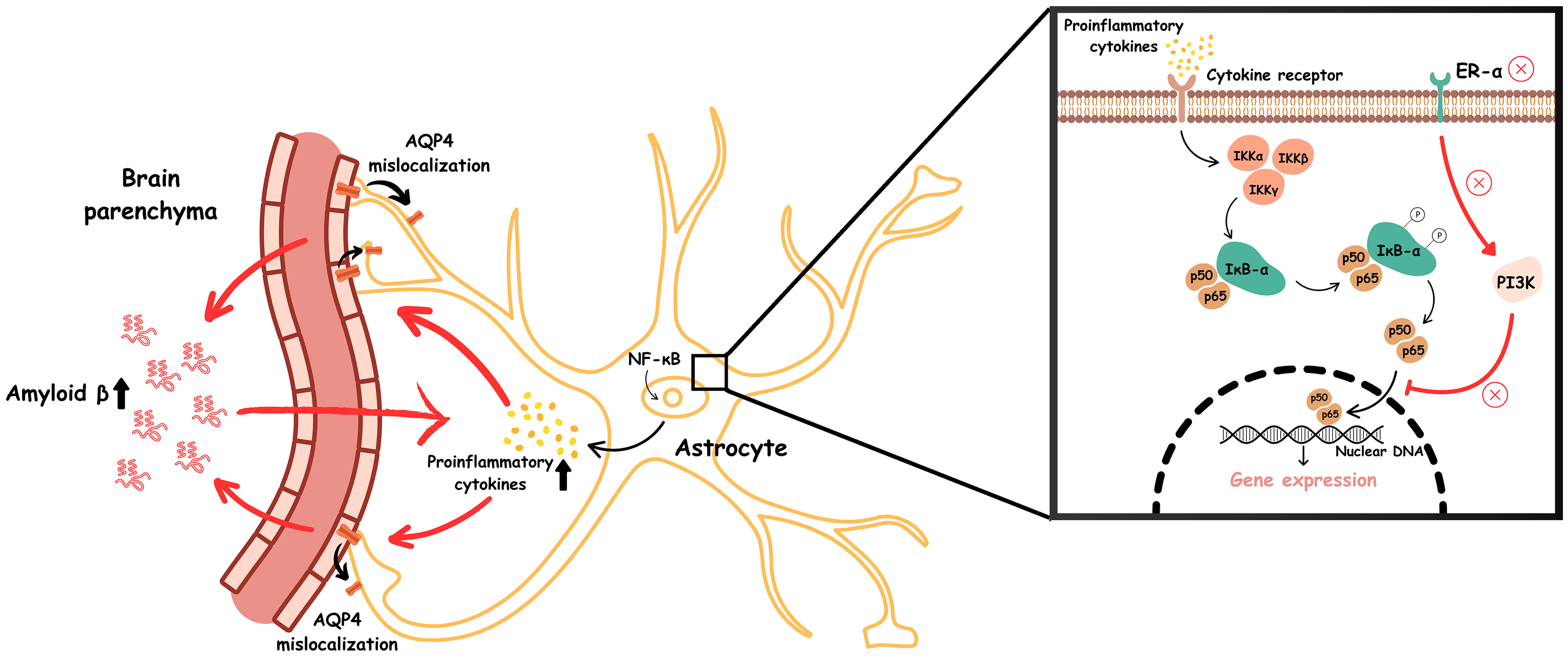
Low estrogen levels result in the loss of ERα-mediated anti-inflammatory signaling. This enables activation of the canonical NF-κB pathway upon the binding of proinflammatory cytokines to their receptors. This, in turn, triggers activation of the IKK complex, composed of IKKα, IKKβ, and NEMO (IKKγ), which phosphorylates IκB-α, marking it for ubiquitin-mediated proteasomal degradation. This degradation releases NF-κB dimers (primarily p50/p65), enabling their nuclear translocation and transcriptional activation of proinflammatory genes. Elevated proinflammatory cytokine production subsequently causes mislocalization of AQP4 away from astrocytic endfeet, impairing glymphatic clearance and promoting the accumulation of toxic proteins such as amyloid-β. This accumulation further amplifies proinflammatory cytokine levels, perpetuating a vicious cycle of neuroinflammation and disrupted protein clearance.
Inflammation grows stronger as we get older, both in the periphery and in the brain. 35 This rise appears in men and women, but it follows different paths in each sex. In women, the years around menopause are a turning point. During this time, the loss of ovarian hormones is matched by sharper immune activity in the blood. The CD4/CD8 T-cell ratio and the numbers of CD4 T cells, B cells, and pro-inflammatory cytokines (IFN-γ, IL-6, IL-8) all climb. 35 Similar changes occur inside the brain. When ovarian hormones fall, microglia in the hippocampus and frontal cortex show more reactivity markers, 35 signaling a state of chronic neuroinflammation.
A similar mechanism is thought to underlie chronic pain and depression, with studies in female mice illustrating how hormonal loss contributes to these outcomes. In both ovariectomised (surgical menopause) and naturally aged mice, the active NF-κB subunit phospho-p65 and the cytokines TNF-α and IL-1β rise in dorsal root ganglia, spinal dorsal horn, and hippocampus. 36 Microglia and astrocytes become activated, and calcitonin gene-related peptide (CGRP) increases in the dorsal horn, boosting C-fiber synaptic transmission. These molecular events translate into chronic pain, memory problems, and depression-like behavior. 36
Elevated cytokine loads are shown to induce AQP4 up-regulation. Nevertheless, they also disrupt its perivascular polarity. 37 Mis-localized AQP4 slows cerebrospinal-interstitial exchange and undermines glymphatic clearance, allowing inflammatory mediators and proteotoxic solutes to accumulate. Thus, chronic neuroinflammation and glymphatic failure reinforce one another in a vicious cycle (Figure 3) that accelerates cognitive decline. 38 Interventions that restore sex-hormone signaling, therefore, represent promising avenues for breaking this loop and improving brain health, particularly in the post-menopausal population.
Conclusions and future directions
Poor clearance of neurotoxic solutes, particularly amyloid-β, has emerged as a central pathological contributor in neurodegenerative disorders such as Alzheimer's disease. Because AQP4 channels at astrocytic end-feet are pivotal for glymphatic water transport, understanding how sex hormones regulate AQP4 is crucial for explaining sex-linked differences in brain edema, fluid homeostasis, and long-term neurodegeneration.
Preclinical evidence to date shows that estrogen and progesterone consistently confer neuroprotection by modulating AQP4 expression or polarity, reducing cerebral edema, and stabilizing the BBB. Testosterone upregulates AQP4 in cultured astrocytes, yet its net impact in vivo remains uncertain. Although none of the reviewed studies directly quantified glymphatic flux, the collective data indicate a hormone-dependent influence on the channels governing glymphatic function.
These findings imply that the sharp decline of estrogen and progesterone after menopause could impair glymphatic clearance, thereby accelerating amyloid accumulation and cognitive decline in women. Consequently, targeting hormone-AQP4 signaling pathways emerges as a promising therapeutic approach to preserve brain fluid balance and slow proteopathic disease progression in a sex-specific manner.
Several limitations temper these findings. Most data come from rodent models, direct measurements of glymphatic flow are lacking, and experimental designs vary widely in hormone dosing and duration. Such heterogeneity restricts the translation of these findings to human physiology and leaves unresolved questions about chronic hormone exposure, aging, and vascular comorbidities.
Future research should integrate real-time in vivo imaging of glymphatic transport with precise manipulation of estradiol, estrone, estriol, progesterone, and testosterone across sexes and age groups. Longitudinal studies linking hormonal status, AQP4 polarity, and cognitive outcomes will clarify causal relationships and guide the development of targeted hormone-replacement or AQP4-modulating therapies.
By revealing how hormones influence AQP4, this study opens the door to sex-specific approaches for maintaining glymphatic function and addressing neurodegenerative diseases. Advancing this line of research could play an important role in supporting healthy brain aging.
Footnotes
Acknowledgements
The authors have no acknowledgments to report.
Author contribution(s)
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.