
Abstract
Background
While APOE4 mutation impacts normal myelination through disruptions of iron and lipid homeostasis, the HFE (homeostatic iron regulatory gene) polymorphism directly participates in white matter (WM) myelination and neuroinflammations processes via modulating iron homeostasis.
Objective
This study investigated effects of HFE polymorphism in APOE4 gene carriers in the context of WM degeneration and neuroinflammation in Alzheimer's disease (AD).
Methods
WM degeneration in AD subjects of APOE4 carriers with and without HFE H63D polymorphism was evaluated and compared with age- and sex-matched cognitive normal (CN) groups using diffusion tensor imaging (DTI) data from the Alzheimer's Disease Neuroimaging Initiative database.
Results
DTI radial and mean diffusivity demonstrated an extensive and precipitous age-related WM degeneration in all the AD groups compared to the CN cohorts. This AD-related WM degeneration, however, was significantly attenuated with HFE H63D polymorphism than that of wildtype along with reduced cognitive decline in the AD group. To link the observed protective effect of HFE H63D polymorphism to WM degeneration and cognitive decline in AD, a mediation model was developed and verified using structure equation modeling. This protective effect of HFE H63D polymorphism on WM is also associated with higher cerebrospinal fluid sTREM2 level.
Conclusions
This is the first genetic-to-imaging study linking HFE polymorphism to WM degeneration and consequentially to cognitive declines in AD. Our data provide original information on the role of iron homeostasis specifically in WM degeneration, which suggests that manipulating iron homeostasis could be incorporated into the overall AD prevention and intervention strategies.
Keywords
Introduction
Tracing back to the genetic predispositions in Alzheimer's disease (AD), genome-wide association studies have demonstrated that the strongest genetic risk factor for sporadic AD is the ε4 allele of the APOE gene (APOE4). 1 The APOE gene encodes for apolipoprotein that plays an important role in lipid transport and homeostasis in the brain. Lipids and fatty acids make up 35–40% of all the molecules in the GM cell bodies and as much as 78% of the axons’ myelin sheaths in the white matter (WM). APOE genetic mutation is likely to impact directly to myelin production, maintenance, and repair. The genetic impact of APOE4 on AD pathogenesis has been shown to be involved in the WM degeneration in addition to the amyloid-β and tau pathology, 2 which highlights the importance of homeostasis of lipid management in WM during aging and associated diseases.
In parallel, AD pathogenesis and neurodegeneration are also linked to the disruptions of homeostasis of iron metabolism in the brain during aging.3,4 Iron homeostasis is accordingly modulated by the HFE (homeostatic iron regulatory) protein that regulates iron uptake by cells. 5 In general, the HFE gene sequence variants (C282Y and H63D polymorphism) are associated with increased cellular iron uptake. But they are also known to modulate inflammatory responses differently, which modifies aging and AD progression.6–11 Our previous experimental data as well as others demonstrated a synergism of HFE polymorphism with APOE4 in AD pathogenesis.7,10 Conversely, recent epidemiology and experimental data revealed that HFE polymorphism plays a significant protective role in neurodegeneration during aging.12–14 Thus, the combined consequences of dysregulation of iron and lipid homeostasis are likely to encompass intermingled biochemical pathways and dynamic processes when responding to neuroinflammations during aging and AD pathogeneses. A recent study using Alzheimer's Disease Neuroimaging Initiative (ADNI) data showed a strong association of ferritin in cerebrospinal fluid (CSF) with preclinical cognitive decline in APOE4 carriers. 15 Thus, the intersect of the gene variants associated with iron and lipids can be an inflection point for the risk factors and a modulator for AD progression.
The focus of AD magnetic resonance imaging (MRI) research has been largely on cortical gray matter (GM) degeneration, especially in medial temporal lobes. With the development of diffusion tensor imaging (DTI) in recent years, the WM changes associated with AD neurodegeneration can be quantified along specific WM tracks. As such, neuroimaging studies have demonstrated that DTI is sensitive for detection of microstructural changes specific to AD in the WM, which is not always detectable by morphological analysis with standard MRI.16–18 Most recently, DTI data indicated that the WM changes may even precede and predict GM volume loss,19,20 making DTI an invaluable tool for assessing corresponding changes in WM during AD neurodegeneration.21,22 As WM have the highest iron content of all cell types in the brain, and that lipids and iron homeostasis are important for myelination by oligodendrocytes, it is rational and imperative to use DTI to determine the impact of WM degeneration in APOE4 and HFE H63D carriers in AD during aging. We hypothesized that HFE H63D polymorphism would reduce WM neurodegeneration in AD APOE4 carriers. To test our hypothesis, the WM integrity of late- and early-myelination regions in bi-genetic mutation (HFE H63D and APOE4) carriers was quantified using DTI and correlated with cognitive scores of study participants. To cast this study into the amyloid/tau/neurodegeneration (AT(N)) framework, 23 CSF biomarkers of these cohorts were collected and incorporated into data analysis. Furthermore, diffusion parameters of WM as mediator for cognitive decline were explored in the context of HFE polymorphism milieu.
Methods
Overview
The data used this manuscript were obtained from the ADNI database (adni.loni.usc.edu). The ADNI was launched in 2003 as a public-private partnership with the primary goal to test whether serial MRI, positron emission tomography (PET), other biological markers, and clinical and neuropsychological assessment can be combined to measure the progression of mild cognitive impairment (MCI) and early AD.
Via Plink software, all the APOE4+ AD patients and cognitively normal (CN) subjects with HFE H63D genetic data were screened. Finally, data from 116 subjects were included in this study, including their DTI, T1-weighted imaging (T1WI), and fluid attenuated inversion recovery (FLAIR) images, clinical diagnosis, neuropsychological assessment and genotyping information APOE4 (rs429358, risk allele: C) and HFE H63D (rs1799945, risk allele: G). AD and CN subjects were further divided into four groups based on genetic information: the HFE H63D and APOE4 allele carrier CN cohort (CNH63D/APOE4+) and AD cohort (ADH63D/APOE4+), the HFE wild type and APOE4 allele carrier CN cohort (CNWT/APOE4+) and AD cohort (ADWT/APOE4+) (Table 1). In addition, to explore the relationship of HFE H63D polymorphism with neuroinflammation, an enlarged cohort without the requirement of DTI data was collected based on availability of CSF sTREM2 within ADNI dataset, which included 52 CN and 70 AD APOE4+ subjects with and without HFE H63D polymorphism (Supplemental Table 2).
Demographics and clinical characteristic of the study cohorts.
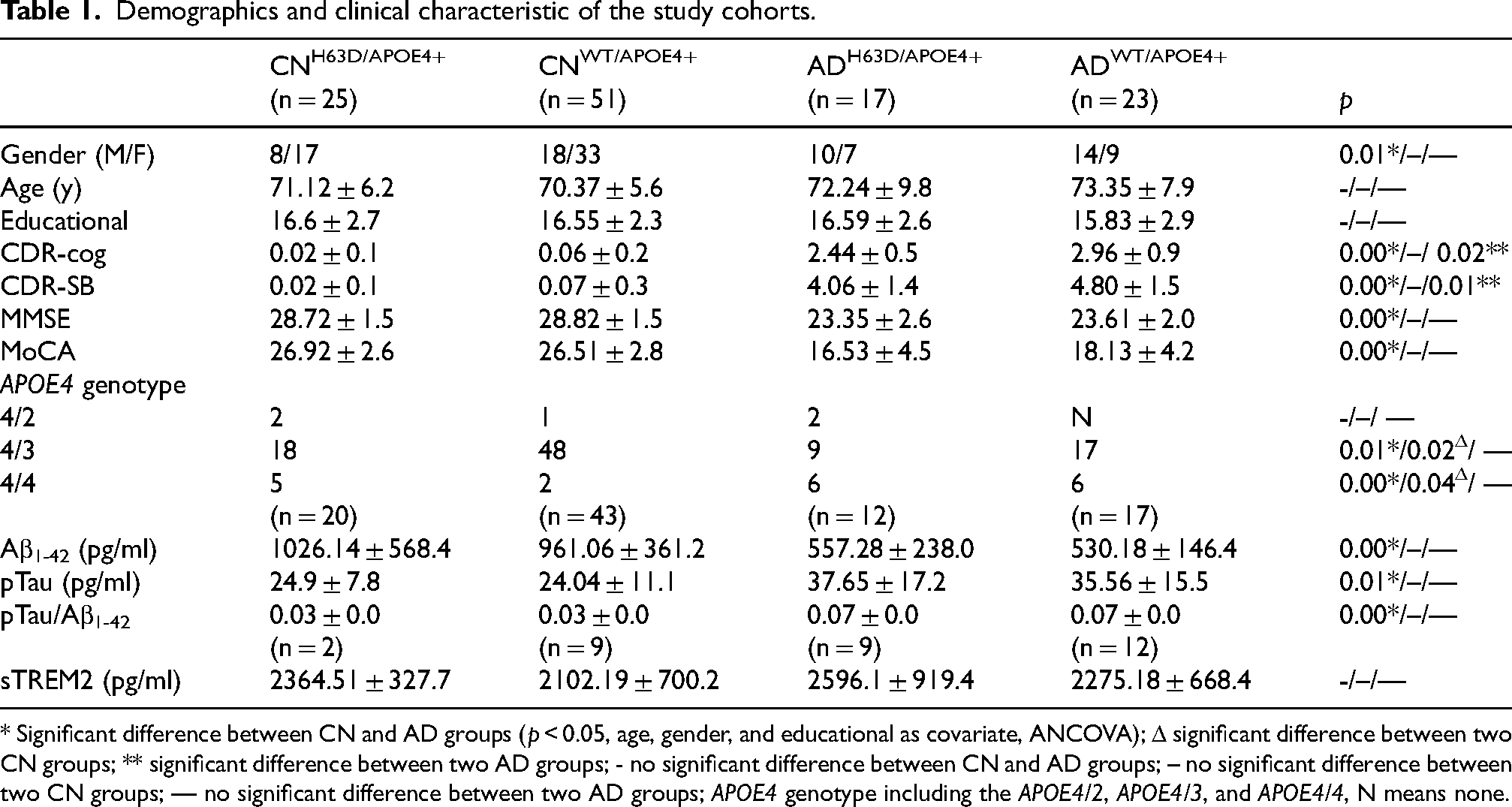
* Significant difference between CN and AD groups (p < 0.05, age, gender, and educational as covariate, ANCOVA); Δ significant difference between two CN groups; ** significant difference between two AD groups; - no significant difference between CN and AD groups; – no significant difference between two CN groups; — no significant difference between two AD groups; APOE4 genotype including the APOE4/2, APOE4/3, and APOE4/4, N means none.
The Montreal Cognitive Assessment (MoCA), Mini-Mental State Examination (MMSE), and Clinical Dementia Rating (CDR) were used as the basic multi-domain cognitive assessments. The CDR assesses three domains of cognition (memory, orientation, judgment/problem solving) and three domains of function (community affairs, home/hobbies, personal care). The CDR scale sum of boxes (CDR-SB) is the total score from all six domains, and CDR-cog is the sum of the memory, orientation, and judgment/problem solving scores. 24
Diffusion tensor imaging protocol
The DTI data were acquired using single-shell acquisition at 3T. Details of scanners parameters were listed in the Supplemental Material (Supplemental Table 1).
DTI data processing
All DTI DICOM images were converted to NIFTI format using dcm2nii 25 and preprocessed using the DTI-studio software (Version 3.0.3). 26 First, the raw DTI images were checked for importing and sorting errors, and looped through in orthogonal views to identify geometric distortions, signal dropouts, subtle system drifts, and missing slices. 27 Based on the diffusion gradient tables, visual inspection of tensor directions in specific brain structures, such as corpus callosum, internal capsule, and brain stem, was performed. Using a standard linear-regression model of diffusion and noise level of 10, fractional anisotropy (FA), mean diffusivity (MD), and radial diffusivity (RD) maps were generated. Using SPM12 (https://www.fil.ion.ucl.ac.uk/spm), the diffusion parameter maps were normalized into the standard Montreal Neurological Institute (MNI) brain template space and smoothed with an 8 × 8 × 8 full-width-at-half-maximum Gaussian kernel.
Previously, we have demonstrated significant reduced FA and increased MD in heavily myelinated white matter regions of H63D-HFE polymorphism carriers. 11 As shown in Figure 1, six major WM structures in the JHU DTI white-matter atlas 28 were chosen as regions of interest (ROI) in the following categories: early-myelination regions (posterior limb of internal capsule (PIC) and cerebral peduncle), late-myelination regions (inferior longitudinal fasciculus (ILF) and superior longitudinal fasciculus, and cognitive related regions (dorsal cingulum and parahippocampal cingulum). Conventionally, the ILF is considered the main component of the sagittal stratum, thus we used the sagittal stratum in the JHU atlas to study the ILF.
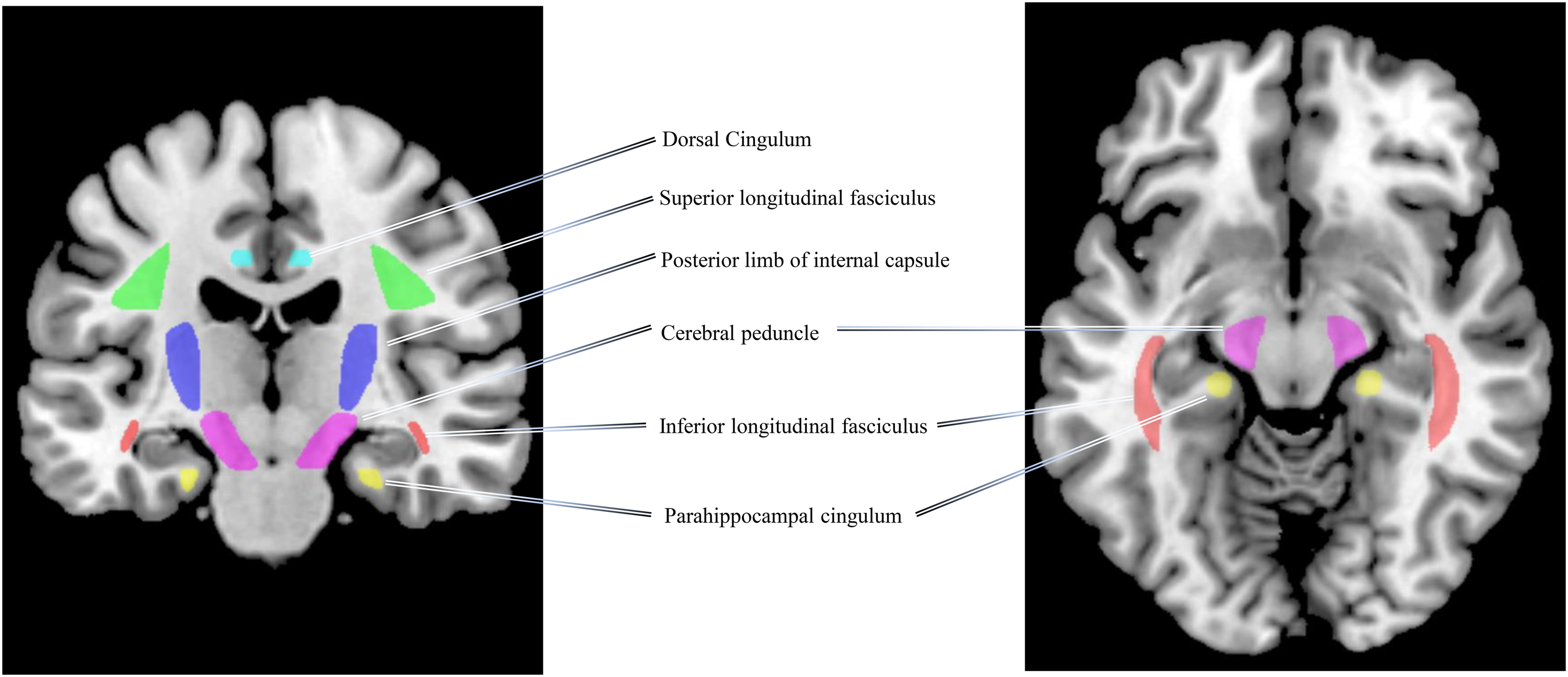
White matter regions of interest in coronal (left) and axial view (right). Early-myelination WM regions: left and right posterior limb of internal capsule (blue), left and right cerebral peduncle (violet); late-myelination WM regions: left and right inferior longitudinal fasciculus (red), left and right superior longitudinal fasciculus (green); and cognitive related WM regions: left and right dorsal cingulum (cyan), and left and right parahippocampal cingulum (yellow).
To remove the effect of free water signals on WM DTI data, 29 the DTI data was co-registered to the FLAIR image and voxels with free water signal were removed from ROI data analysis. There was no significant difference in the number of voxels with free water signal in each WM ROI among the four study cohorts (ANOVA, no significant difference).
Morphological data processing
The whole brain T1-weighted MRI images were preprocessed with SPM12 following the standard procedure of segmentation, normalization into MNI space, and then spatially smoothed using an 8-mm full-width-at-half-maximum Gaussian kernel. Voxel-based morphometry (VBM) analysis of brain WM was conducted with SPM12.
CSF biomarkers collection of sTREM2, amyloid-β1 - 42, and pTau
The CSF biomarker data available in the ADNI database from the selected subjects were also obtained. All the CSF samples were collected from each subject within four weeks when the DTI scans were performed. The CSF biomarker measurements were performed by the ADNI Biomarker Core laboratory at the University of Pennsylvania Medical Center. The data ranges (lower technical limit to upper technical limit) of these assays were: 200 to 1700 pg/ml for Elecsys Aβ1–42, and 8 to 120 pg/ml for pTau181. The CSF sTREM2 assay is based on the WU platform and has been comprehensively described previously.30,31 A detailed description of the CSF sTREM2 measurements in the ADNI samples, as well as the original data, can be downloaded from the LONI Image and Data Archive (https://ida.loni.usc.edu).
Statistical analysis
Differences in age, sex, educational years and APOE4 genotype across groups were assessed using Wilcoxon rank test and Pearson's Chi-squared test. Cognitive performance and biomarkers’ differences between groups were assessed using ANCOVA with age as covariance. Statistical significance level was set at p < 0.05 (Table 1).
Aging effect on diffusion parameters of WM was evaluated via linear regression. With significant aging effect, group-wise ROI-based analyses of diffusion parameters, i.e., MD, RD, and FA of predefined WM structures (Figure 1) were conducted with age as a covariate to examine the influence of HFE H63D on WM degeneration in AD via ANOVA. When there was a significant inhomogeneity of variance between groups, diffusion parameters were log-transformed, and post hoc tests were conducted. Statistical insignificant threshold was set at p = 0.05 with multiple comparison correction. Correlations between diffusion parameters of WM structures in AD subjects and cognitive function were evaluated using linear regression with age as a covariate.
To explore the directed correlations of HFE polymorphism to DTI and cognitive decline Structural Equation Modeling (SEM) was conducted using the lavaan package in R. 32
To examine the influence of HFE H63D on brain atrophy in AD, cross-cohort VBM analysis of GM and WM was conducted using ANOVA with age and intracranial volume (ICV) as covariates. Statistical significance level was set at p < 0.05 (family-wise-error corrected) with an extent threshold of 100 voxels.
Results
Study cohort characteristics
The demographics and clinical characteristics and APOE4 genotype of the four study cohorts are listed in Table 1. In each cohort, the number (n) of the subjects with CSF pathological markers (Aβ1-42 and pTau) and CSF inflammatory marker (sTREM2, pg/ml) are smaller due to the availability of CSF data for each of our cohort within the ADNI database. According to the ATN framework criteria (CSF Aβ1-42 cut-offs of 977 pg/ml, <977 pg/ml as “A+”; CSF pTau cut-offs of 27 pg/ml, ≥27 pg/ml as “T+”; CSF pTau/Aβ1-42 cut-offs of 0.025, ≥0.025 as “A + T+”),23,33 the AD subjects in this study were pathologically confirmed within the ATN framework, while the CN subjects had mixed pathological profiles. Few subjects carried the APOE2 allele, while the majority exhibited either the APOE3/4 or APOE4/4 genotype. This distribution aligns with that observed in the general population. 34 The APOE3/4 ratio is significantly higher in the CN cohort compared to the AD cohort, whereas the APOE4/4 ratio is significantly lower in the CN cohort than in the AD cohort. This is in accordance with established findings that APOE3 serves as a neutral factor for AD, whereas APOE4 acts as a risk factor for AD, particularly in the case of APOE4 homozygous carriers. 35
Association of HFE H63D polymorphism with diffusion parameters in WM
The average DTI metrics, RD, MD, and FA of the four groups are shown in Figure 2. A significant trend of augmented RD and MD was found in AD subjects in most WM structures studied, demonstrating a widespread WM degeneration in AD. The corresponding FA in AD subjects appeared to decrease accordingly, but to a less degree. RD has been shown to be more sensitive in detecting WM changes than FA and MD by the previous studies. 36 Most interestingly, the comparison between the two AD groups revealed that the increased RD and MD in the ADH63D/APOE4+ group were less than those in ADWT/APOE4+, significantly in the bilateral ILF and right PIC (ANOVA, age as a covariate, multiple comparison corrected, p threshold is 0.05). These results demonstrated that the WM degeneration was lessened with HFE H63D polymorphism in the AD APOE4+ carriers. To further investigate this effect of HFE H63D polymorphism on WM neurodegeneration in AD, we focused on the bilateral ILF where the WM degeneration appeared more prominent.
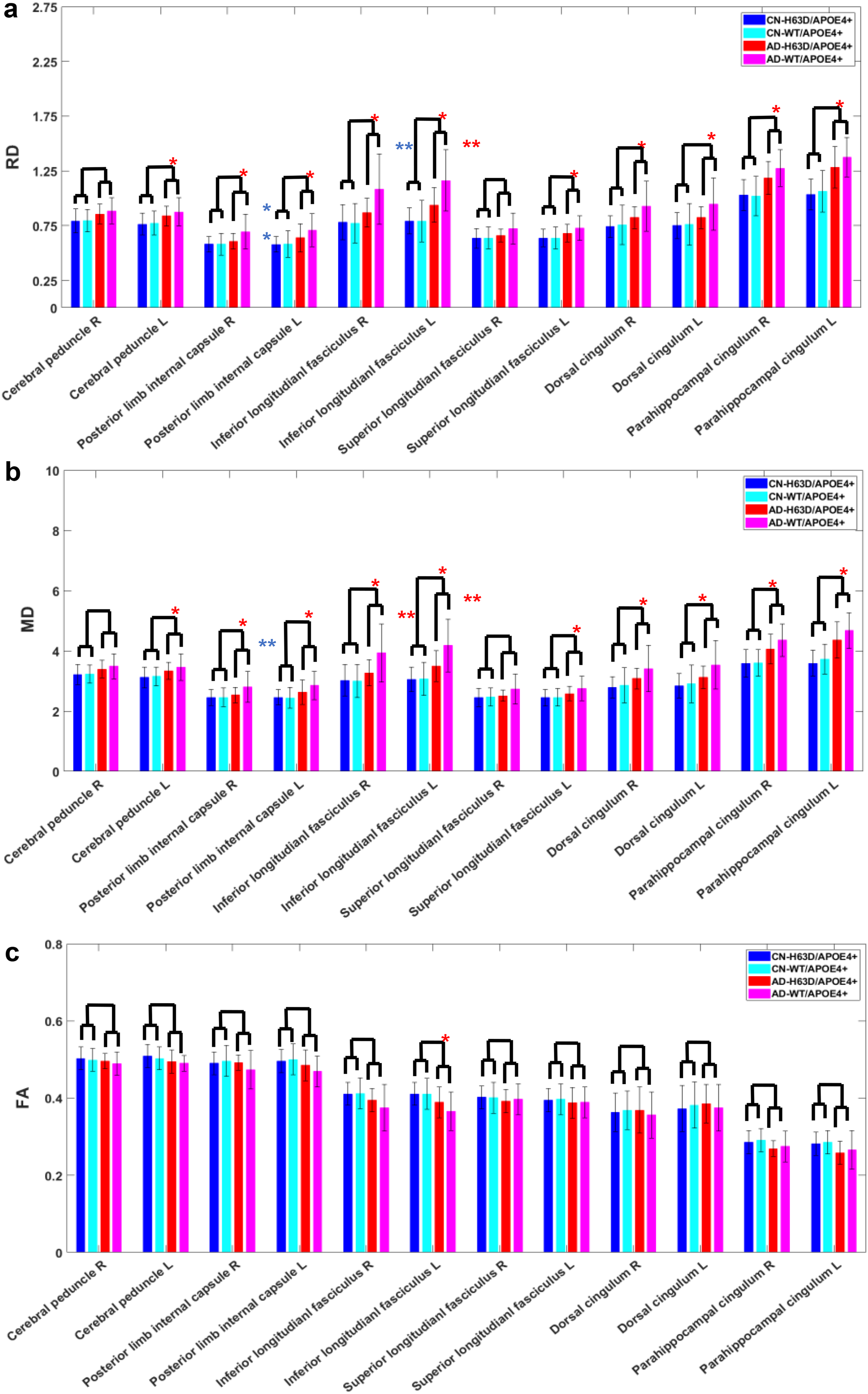
Association of HFE H63D mutation with DTI parameters in WM. Augmented RD (A) and MD (B) in the AD groups, significantly in most WM structures, indicating a pervasive WM degeneration. FA (C) in AD was decreased accordingly but to a less degree. Between the two AD groups, the increases in RD and MD in ADH63D/APOE4+ group were less than those in ADWT/APOE4+ group, significantly in bilateral inferior longitudinal fasciculus and posterior limb of right internal capsule (p value in figure). *significant difference between AD and CN groups (p < 0.05); **significant difference between two AD groups (age as a covariate, multiple comparison corrected, p < 0.05); **significant difference between two AD groups (age as a covariate, without multiple comparison correction, p < 0.05, with multiple comparison correction, p = 0.12).
HFE H63D polymorphism's effect on age-related WM neurodegeneration
To investigate this finding further, we examined age-dependence of RD and MD of bilateral ILF in the AD and CN cohorts. As shown in Figure 3, there was significant positive correlation between diffusivity and age in all four groups: higher level of WM degeneration with greater age. The slopes or the rate of change with age in both RD and MD in the ADH63D/APOE4+ group were lower than those of ADWT/APOE4+ group but similar to the two CN groups. Furthermore, the interaction effect between HFE H63D genotype and age in RD and MD was significant in ILF_R (pinter = 0.022 and 0.023, respectively), which suggested that HFE H63D significantly influenced aging differences between the two AD groups. Such interaction effect was not reached to a significant level in the left ILF nor observed in the CN groups. These results imply that the effects of the HFE H63D polymorphism counteracted age-related WM degeneration. Accordingly, FA of IFL was negatively correlated with age. Although the slope for ADH63D/APOE4+ was reduced compared to that in the ADWT/APOE4+ group (in the bilateral ILF), no significant interaction between age and H63D polymorphism on FA was observed in our cohorts (pinter = 0.905 and 0.249) (Supplemental Figure 1).
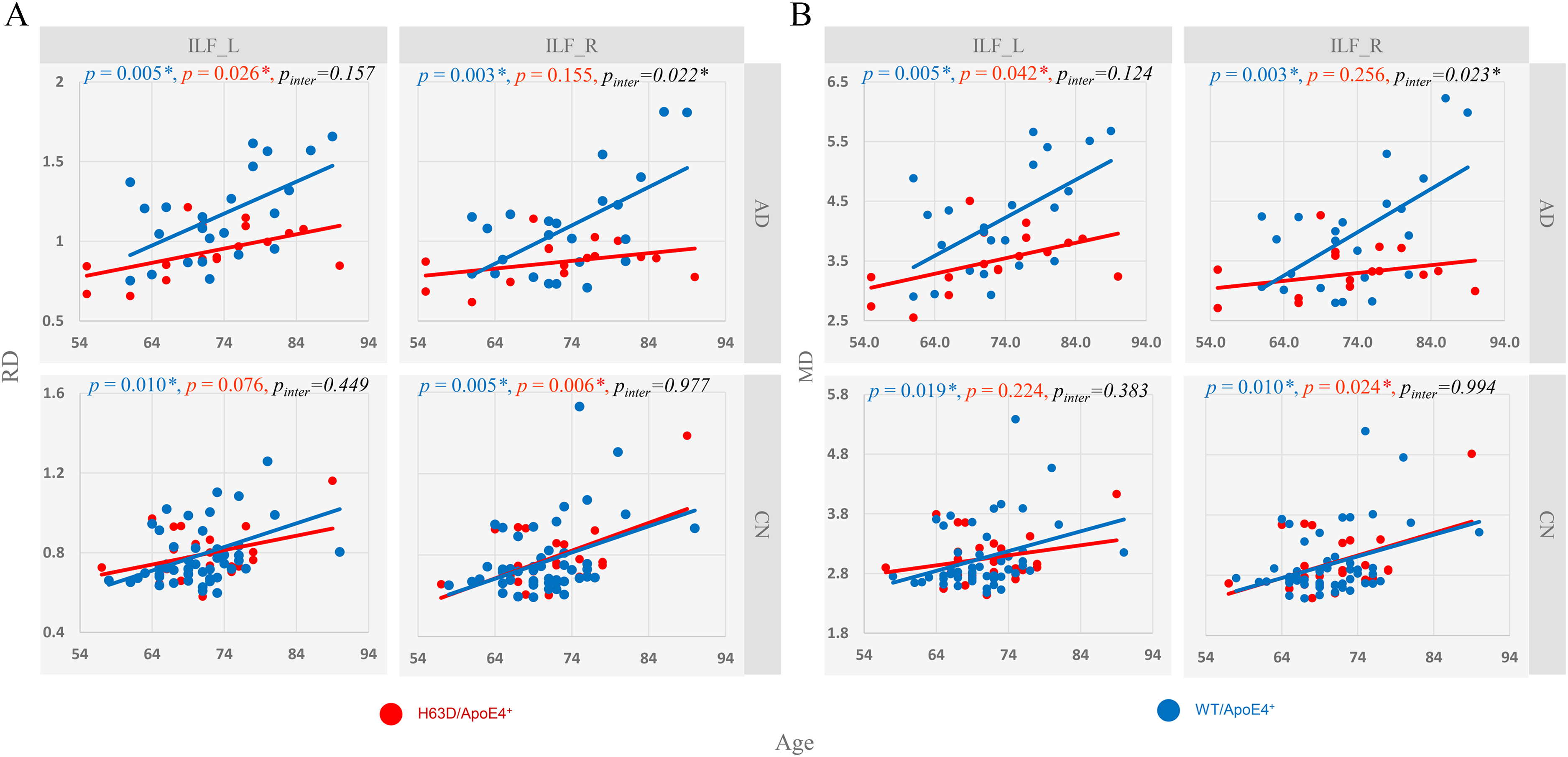
Age dependence of RD (A) and MD (B) of bilateral inferior longitudinal fasciculus (ILF_R and ILF_L) affected by HEF H63D polymorphism. The slopes of linear increase of RD and MD with age in the ADH63D/APOE4+ group were lower than those of the ADWT/APOE4+ group, but similar to the two CN groups. Note: Linear regression was used for association between age and RD or MD. p is for the correlation between age and outcomes (RD or MD) in ADWT/APOE4+ and CNWT/APOE4+ groups, p for the correlation in ADH63D/APOE4+ and CNH63D/APOE4+ groups, and p inter is for the interaction effect between age and HFE H63D genotype category on outcomes. For the AD cohorts, a significant interaction between age and HFE H63D on RD and MD was found in ILF_R (p < 0.05).
Association of WM neurodegeneration and cognitive impairment
As demonstrated above, RD exhibited the largest differences among the three DTI metrics in detecting WM degeneration between CN and AD, especially between the two AD cohorts. Its associations with cognitive scores were, thus, calculated in the AD subjects. As shown in Figure 4, RD of ILF, as a proxy of WM neurodegeneration, was significantly correlated with CDR-cog, MMSE and MoCA among all the AD subjects (p ≤ 0.039, age as covariate). In addition, RD of left dorsal cingulum and bilateral parahippocampal cingulum as shown in Table 2 were significantly correlated with cognitive scores. The cingulum and its parahippocampal component are composed of WM fibers that connect various regions of the limbic system, including the cingulate gyrus, parahippocampal isocortex, and medial temporal lobe. The dorsal cingulum connects the brain areas in the default-mode network involving in cognition, while the paprahippocampal cingulum plays a role in memory and spatial orientation. 37 And degeneration in cingulum also concurs with damages the hippocampus in MCI and AD.38,39 Along with results in Figure 4, our data demonstrated that WM degeneration, particularly in the bilateral inferior longitudinal fasciculus, could mediate the cognitive impairment in AD during aging.
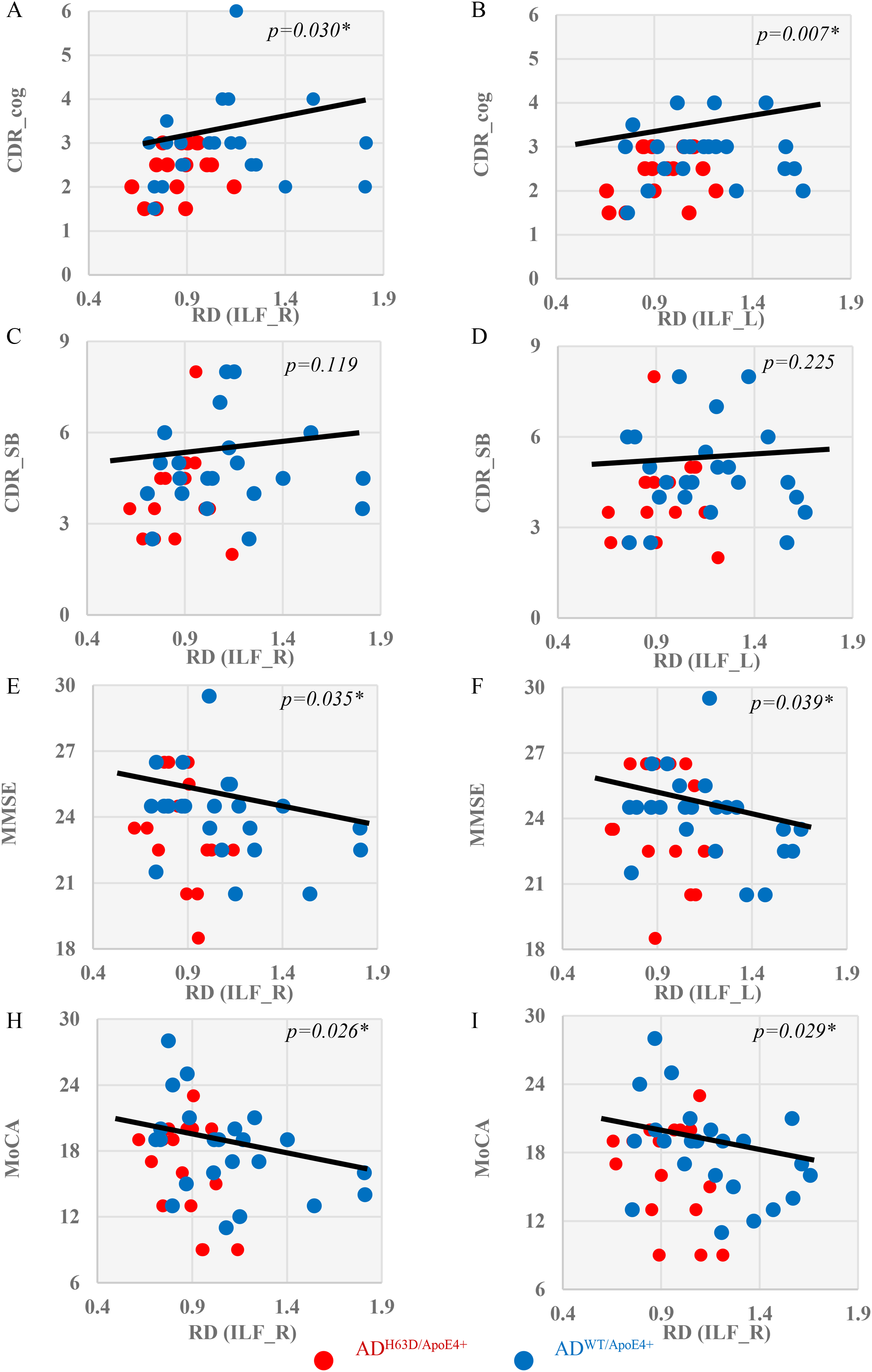
Association of white matter neurodegeneration and cognitive impairment measures. Significant correlations (p < 0.05, age as covariate) were found between RD of inferior longitudinal fasciculus (ILF) with CDR-cog (A, B), CDR-SB (C, D), MMSE (E, F) and MoCA scores (H, I) in the AD cohorts.
p-values for correlations of RD of the WM ROIs with cognitive measurements (outcome) in all AD subjects.
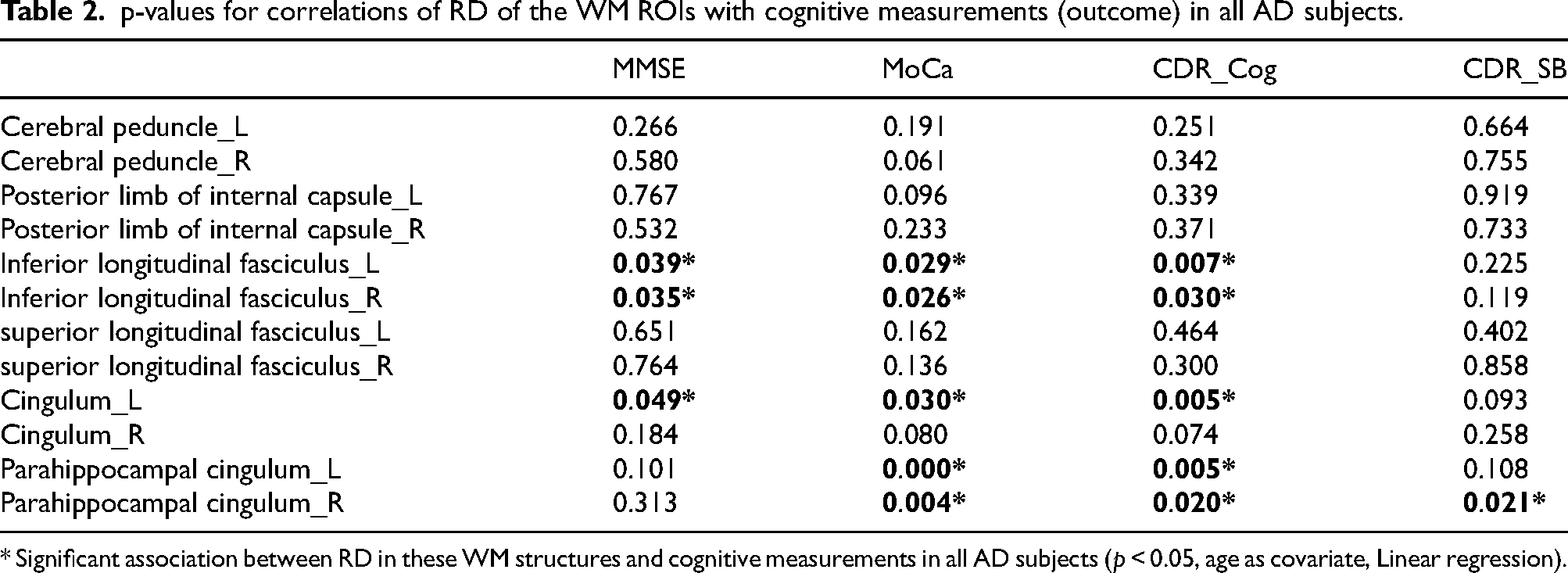
* Significant association between RD in these WM structures and cognitive measurements in all AD subjects (p < 0.05, age as covariate, Linear regression).
WM neurodegeneration mediates the effect of HFE H63D polymorphism on and cognitive decline in AD
In order to piece the above univariate analyses together to produce a coherent picture of the impact of HFE polymorphism on AD cognitive declines, a mediation analysis was conducted with SEM built upon our data set and current literatures. Figure 5 shows a plausible model of directed (causative) effects of HFE polymorphism on cognitive declines mediated by WM degeneration in AD. In this model, age and HFE H63D polymorphism are independent variables, while RD_ILF is a latent variable, as a mediator derived from RDs of ILF_L and ILF_R. While age exerts a positive effect directing to bilateral RD_ILF (p < 0.001) as anticipated, HFE H63D polymorphism applied a negative effect on RD_LF (p < 0.001). Subsequently, RD_ILF exerted a negative effect on MoCA (p = 0.031). This result suggests that a lower WM degeneration leads to a higher MoCA score. This modeling fits our overall data well as evidenced by a significant Chi-square test (χ 2 (4) = 10.972, p = 0.027).40–42 The same SEM analyses using MMSE and CDR_cog as dependent outcomes yielded similar but non-significant results (χ 2 (4) = 5.461 & 6.082, p = 0.243 & 0.193, respectively) as shown in Supplemental Figures 4 and 5. Taken together, the SEM results confirmed that HFE H63D polymorphism exerts a protective effect on age-related cognitive decline in AD APOE4+ carriers mediated by reducing WM degeneration. Thus, the interactions between HFE polymorphism and age on the WM degeneration in the AD cohorts as shown in Figure 3 manifested a profound impact on AD cognitive decline.
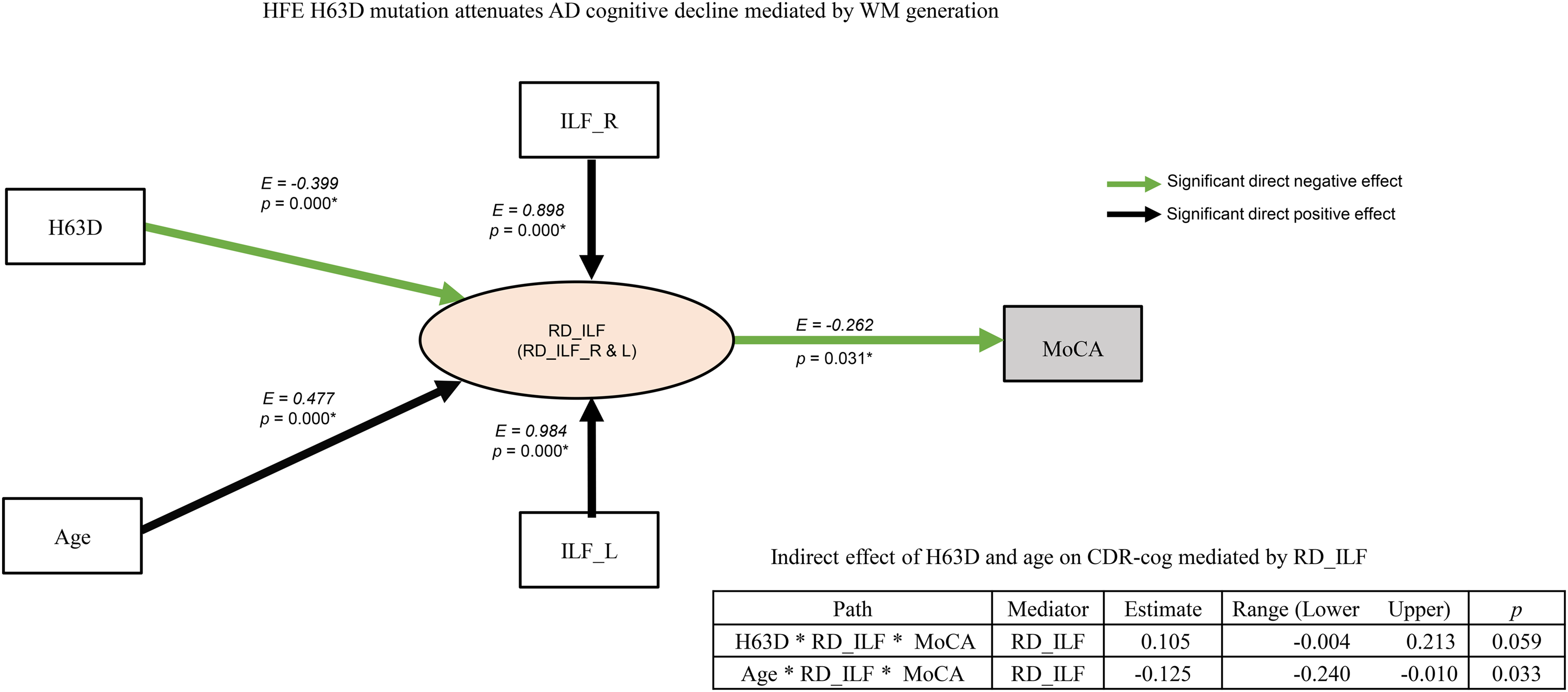
The SEM model of the mediation effect of HFE H63D genotype and age on MoCA in the AD cohorts. In this model, HFE H63D mutation and age are independent variables. The lines with arrowhead indicate causative or directed effect on the dependent variables. The E value listed next to each line indicates the estimate of path coefficients between each pair of variables. RD_ILF is a latent variable derived from the summary of RD_ILF_L and RD_ILF_R (EL = 0.99, ER = 0.89, p < 0.001). Age exerts a negative effect on MoCA (inset table), which is mediated via its direct and positive effect on RD_ILF (solid black line). HFE H63D mutation positively influences MoCA indirectly (inset table), which is mediated via a direct but significantly negative effect on RD_ILF. The SEM model suggests a significant protective effect of HFE H63D mutation on AD cognitive decline via attenuated WM degeneration (Chi-square test (χ2 (4) = 10.972, p = 0.027).
Neuroinflammation and degeneration in WM
Since iron is closely involved in signaling pathways of inflammation, the potential protective mechanisms of HFE polymorphism on WM degeneration was further explored using CSF neuroinflammatory marker, sTREM2.43–45 As seen in Table 1, sTREM2 is higher in the ADH63D/APOE4+ group than that of ADWT/APOE4+ group, which became statistically significant (p ≤ 0.01) with an enlarged AD cohort based on all the CSF sTREM2 data available in ADNI database as shown in Supplemental Table 2. As demonstrated in Figure 6, the level of CSF sTREM2 is negatively correlated to CDR-cog with the AD cohorts together (p = 0.023). This result suggested that the protective effect of HFE H63D polymorphism in Figure 5 was likely due to the contributions of maintaining positive neuroinflammatory activities in sTREM2, which is constant with a previous study showed that sTREM2 ameliorates pathological phenotypes by modulating microglial functions. 46
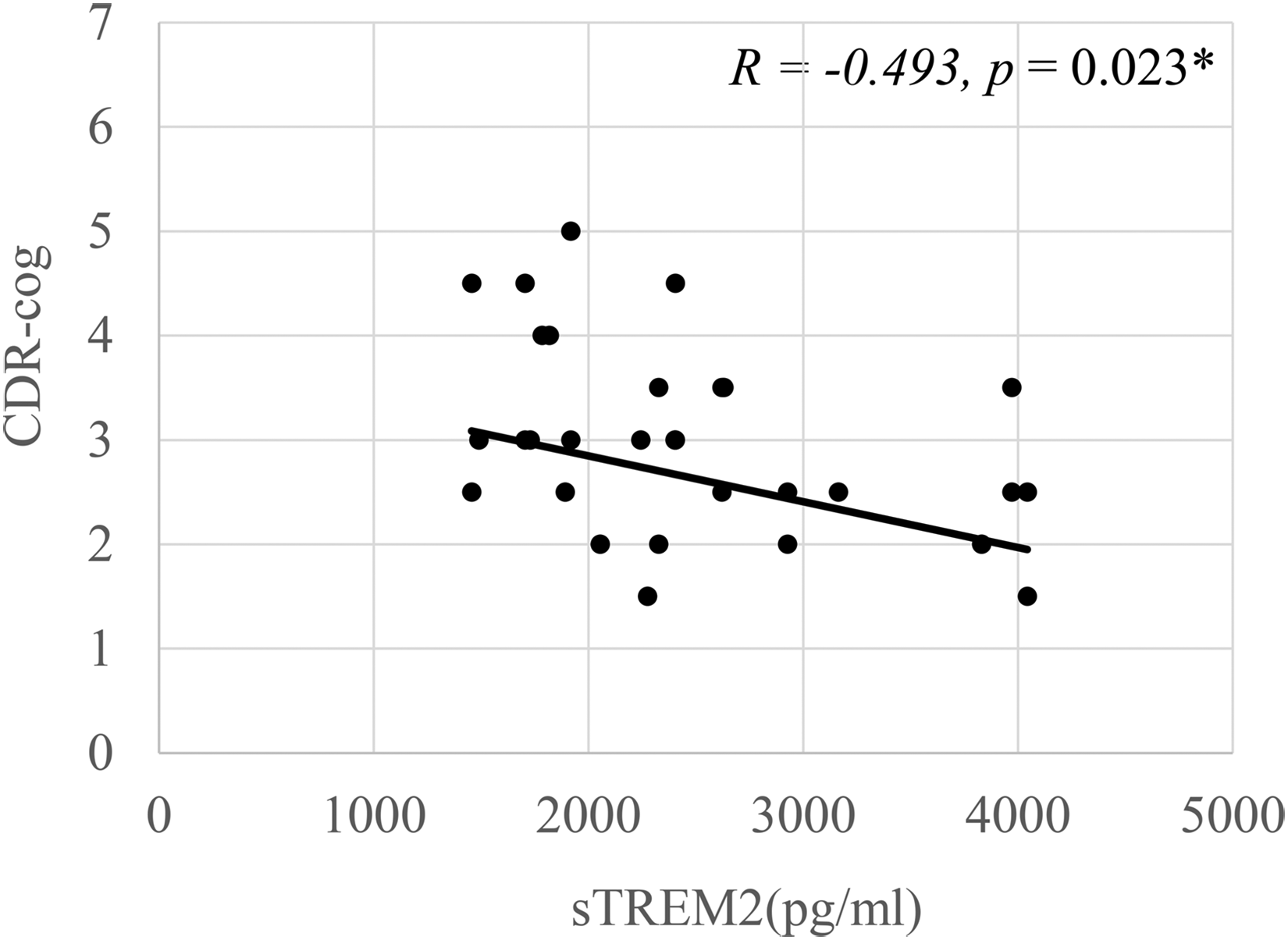
Correlation analysis between CSF sTREM2 and cognitive decline in the AD cohort. Significant negative correlations were found between sTREM2 and CDR-cog in the AD cohorts (Linear regression, with age and gender as covariates, p ≤ 0.023).
Morphology analysis
Compared to the CN subjects, widespread GM atrophy was observed in AD subjects, especially, in bilateral temporal lobe, orbitofrontal cortex, hippocampus, amygdala, and posterior cingulate (ANOVA with age and ICV as covariates, family-wise-error corrected, p < 0.05, extent threshold = 100 voxels, Supplemental Figure 2). Significant WM atrophy was observed in AD in bilateral inferior temporal lobe, medial temporal lobe, frontal lobe, fornix, and left orbitofrontal subcortical area with the same statistical threshold (Supplemental Figure 3). There was no significant difference, however, in GM or WM volume between the two AD groups or between the two CN groups.
Discussion
It is difficult to link the AD genetic risk factors to their manifestations during AD progression at the gross brain anatomy level that can be detected by neuroimaging due to various comorbid pathologies during aging. This study attempted to elucidate how the genetic risk factor of HFE H63D polymorphism impact on WM degeneration in AD progression in the common genetic background APOE4 mutation. Both ROI and voxel-based analyses demonstrated a widespread WM degeneration in AD subjects with APOE4 mutation compared to age-matched APOE4+ CN groups corroborating previous studies.21,22 The most important finding of our study, however, was the significant attenuation of WM degeneration along specific WM tracks in bi-genetic carriers ADH63D/APOE4+ compared to ADWT/ APOE4+ (Figure 2). In contrast, little difference was observed in GM atrophy between the two AD cohorts. The attenuated WM degeneration in ADH63D/APOE4+ carriers interacted with age (Figure 3). The increases in RD along several major WM tracks were significantly correlated with cognitive decline in AD (Figure 4 and Table 2). As shown by our mediation analysis with SEM, the effect of HFE H63D polymorphism has a significant but negative effect on WM degeneration (Figure 5), leading to an overall negative effect on cognitive decline. Additionally, this attenuated cognitive decline could be attributed to the increased activity of sTREM2 (Figure 6, Table 1, and Supplemental Table 2), which likely involve in offsetting the effect neuroinflammation on WM degeneration and aging. Taken together, our data suggest that HFE H63D polymorphism provides a ‘protective’ effect in APOE4+ AD.
Our study established a specific link between the HFE genetic mutation and in vivo measurement of WM degeneration, which acts as a mediator to the observed cognitive decline in AD. The HFE and APOE genes encode proteins responsible for iron and lipid homeostasis respectively, and both proteins are intimately involved in WM myelination and demyelination processes. Thus, to understand the mechanism and significance of our findings, it is necessary to examine the role of the HFE polymorphism in brain iron management and its influence of APOE4 mutation on lipid homeostasis—both in the context of AD-related WM degeneration and neuroinflammation.
Interconnection of lipid and iron in WM
Our data and those of others implicated abnormalities in WM in AD during aging.16,47–51 Recent studies suggest that problems with brain cells’ ability to process lipids, particularly for myelination, may play a key role in AD and related diseases.17,47,52,53 Myelin in the brain consists of lipids and is primary produced by oligodendrocytes cells. In addition to lipids, iron is directly involved in myelin production as a required co-factor for cholesterol and lipid biosynthesis, and indirectly involved in the oxidative metabolisms. As such, oligodendrocytes have the highest iron content among all brain cell types, which increases with age and is even higher in AD. 54 During myelination, large amounts of lipids and membrane must be processed and produced by oligodendrocytes, which require 2 to 3-fold higher energy levels than other cell types in the brain. On the other hand, oligodendrocytes at all stages of their differentiation, compared to other glial cells, contain smaller amounts of antioxidant agents such as glutathione peroxidase and only half of the glutathione reductase activity. 55 Thus, a high metabolic demand and iron content with a low antioxidant level make oligodendrocytes one of the most vulnerable cell classes to oxidative stress in the central nervous system. Myelination processes are, therefore, metabolically demanding, making oligodendrocytes particularly vulnerable to insults of hypoperfusion, excitotoxicity, and oxidative stress. These insults become more widespread and chronic during aging and are exacerbated during AD stage. Thus, the consequence of APOE4 mutation on WM degeneration in AD is direct and profound, which is coupled closely with changes in iron metabolism and oxidative stress responses brought about by HFE polymorphisms. It has been hypothesized that age-associated myelin breakdown leads to iron release from oligodendrocytes and consequentially promotes Aβ oligomerization in the parenchyma.56–58
Impact of APOE4 and HFE polymorphism on WM degeneration
It is well established that, APOE4 directly induces ferritin in CSF, 15 which serves as a precedent for the current HFE-specific study. As demonstrated here, the HFE mutation of the iron regulation protein with concurred APOE4 mutation could strongly impact WM degeneration in AD subjects. APOE4 gene has been identified as the strongest genetic risk factor for AD; possessing one and homozygous APOE4 allele increases the risk of developing AD by approximately 3–7 times and 12 times, respectively. 59 The mechanisms for such a strong effect in increasing AD's risk has been studied extensively and yet completely understood. Based on the fact that ApoE protein plays an important role in transportation and recycling of lipids in the brain, its genetic mutation is likely to play a more direct role in WM for myelin maintenance and repair60–62 The HFE gene, on the other hand, encodes membrane protein required for regulation of cellular iron uptake.5,63 The H63D missense mutation is one of the most common genetic polymorphism in Caucasians (HFE H63D, rs1799945) being present in 12–15% of the population normally and increased to nearly one-third of the patients with neurodegenerative diseases. Thus, polymorphism within the HFE protein gene have been interrogated for increased risk of developing a number of neurodegenerative disorders such as amyotrophic lateral sclerosis, Parkinson's disease, and AD, for which it is believed to be due to the downstream effects in iron dyshomeostasis and increased oxidative stress.6,7,10,12,64 This study confirms the significant role of iron mis-regulation as a major risk factor for the onset of AD corroborating well with the fact that the Alzheimer's amyloid precursor protein is controlled by Iron regulatory protein 1 (IRP1) and the level of translation of its message. 65 Recent animal studies, however, have suggested that HFE H63D polymorphism significantly impacts a number of critical macrophage functions. Specifically, fundamental activities such as proliferation in response to iron exposure, L-ferritin expression in response to iron loading, secretion of BMP6 and cytokines, and migration and phagocytic activity. 66 Vulnerability of the WM to AD pathogenesis likely also involves the inflammation-mediated neurodegeneration mechanism, which is associated with activation and iron sequestration of microglia. Under normal conditions, microglia activation is responsible for removal of toxins and myelin debris from the brain, which is crucial for re-myelination.67,68 In responses to WM injuries by AD pathologies, iron sequestration in microglia could impact AD pathogeneses differently with HFE polymorphism.69–72 At clinical AD stage, neuroinflammation becomes more widespread and aggravated, which could lead to a lower iron in plasma and iron sequestration in microglia via IL-6 mediated hepcidin induction.57,58 Thus, IL-6 mediated hepcidin pathway could play an important role on inflammation-related iron-modulation, feeding the vicious AD oxidative-stress-neuroinflammation-degeneration cascade.66,73–75 As described by Adeniyi et al., such WM injuries in the aging brain appeared to be highly susceptible to microglial degeneration from ferroptosis because of the compromised ability of senescent microglia responding to the myelin debris due to WM injuries. 76 Since ferroptosis is a type of programmed cell death, which is heavily dependent of iron, our data suggesting an interesting hypothetical mechanism that the iron sensing systems in HFE H63D carriers could be attenuated during this process, which could alleviate the ferroptosis and subsequently slowdown the WM neurodegeneration. This mechanism is also well corroborated with recent literatures. Most noteworthy, the temporal profile of microglial activation in AD elucidated by Fan et al. is presumably consisted of two peaks of microglial activation in the AD progression trajectory: an early protective peak in MCI stage and a late cidal pro-inflammatory peak as amyloid clearance fails and AD progresses. 46 During this process, the iron sensing systems is attenuated in HFE H63D carriers, which could alleviate the neuroinflammation cascade and subsequently slow down the AD neurodegeneration. In our cohorts with HFE H63D polymorphism, the late pro-inflammatory activation of microglia seemed reduced at AD stage and thereby slowing down neuroinflammation cascade, and subsequently the AD neurodegeneration. The fact of the alleviated CSF sTREM2 in the AD cohort with HFE H63D mutation compared to that of the wide type (Table 1 and Supplemental Table 2), and its negative correlation with cognitive decline (Figure 6) support our hypothesis. The impact of HFE polymorphism on WM degeneration is likely through the attenuation of microglial ferroptosis.
DTI in WM and morphology in GM measurements
Our DTI analysis demonstrated an extensive and precipitous age-related WM degeneration in ADWT/APOE4+ carriers, which was significantly lessened in the ADH63D/APOE4+ subjects with reduced cognitive decline. The accompanying WM and GM volumetric measurements showed significant differences between the AD and CN, consistent with previous findings. However, the morphology measures failed to show differences between the two genetically variant AD cohorts. These results seem to suggest that WM is more responsive to HFE H63D polymorphism, and consequentially, DTI measures along specific WM tracks are more sensitive in detecting cognitive-related neurodegenerations during AD. For example, WM tracks such as the ILF and PIC, are presumably connected to temporoparietal association cortical areas where cognitive functions are profoundly dependent of. Thus, their catastrophic dysconnectivity could lead to clinical presentation of dementia. In addition, WM tracks in our AD cohort overlaps topographically with known AD-related cerebral blood perfusion deficits in temporoparietal cortical areas. These results also suggest that DTI measures (e.g., RD) in the WM tracks connecting the temporoparietal association cortex may offer pathologic measures related to cognition for AD staging, which could be important for developing therapeutics.
Limitations and future directions
The HFE polymorphism contribution to AD remains paradoxical despite extensive studies using various transgenic animal models. 8 Validation of animal study findings in humans with clinical patients has been limited because it would require a longitudinal study with a large sample size. Our study only examined HFE genetic impact during AD stage cross-sectionally with the available data. In addition to H63D, the other prevalent and well-studied HFE gene variant is C282Y. We did not include this gene variants into our study because only few AD subjects with concurred C282Y and APOE4 mutations could be found in the ADNI database. In addition to its relatively lower prevalence (5.4%) compared to H63D mutation (13.5%) in the US population, the other reason for such a low incident of HFE C282Y mutation in AD could be also related to its protective effect on AD. An epidemiology study by Tisato et al. showed that possessing one C282Y allele could extinguish the APOE4 allele associated risk completely. 13 Another compounding contributor could be that people with C282Y mutation are at a higher risk of hemochromatosis, which would preclude their chance for being recruited into the ADNI study.
The findings of this gene-to-imaging study highlight the importance of iron regulation in responding to neuroinflammation in WM during AD progression. Thus, to pursue this question further, several limitations need to be addressed in future studies. First, the cohorts’ classifications in ADNI were based on clinical assessments since we did not have a complete amyloid-β PET data set as ADNI project started (2004) before ATN framework was established in 2018. 23 We, however, collected CSF biomarker data from the majority of our subjects (Table 1) that helped confirmation of the AD diagnoses for this study. As more data within the ATN framework become available in the coming years, we plan to conduct further analyses with larger cohorts. Second, it would require a much greater sample size to investigate genetic effects of HFE mutation alone on AD under the shadow of overwhelming effect of APOE4 mutation. By selectively using APOE4 mutation as a common genetic background, the effect of HFE polymorphism as a disease modifier for AD emerged. As demonstrated in this study, the interactions between the two genetic mutations are highlighted in the WM degeneration during AD, which is unlikely accidental. In order to untangle the interrelated effects of iron and lipid homeostasis on AD pathogeneses further, AD and MCI subjects of none APOE4 carriers (homozygous and heterozygous with APOE3 and APOE2) with HFE H63D polymorphism should be investigated longitudinally with comprehensive neuroinflammation biomarkers. This would allow us to isolate the genetic impact of HFE H63D polymorphism on AD progression and generalize our findings. Thirdly, to test our hypothetical mechanism linking iron-lipids hemostasis to WM degeneration, animal models with HFE mutation with other AD genetic risk factors must be developed to investigate the underlining biochemical processes associated with iron and lipoproteins along with neuroinflammation biomarkers.
Conclusions
This is the first gene-to-imaging study linking HFE polymorphism to WM degeneration, and consequentially, to cognitive declines in AD bi-genetic HFE H63D and APOE4 carriers. We demonstrated a protective effect of HFE H63D polymorphism on AD WM degeneration in relation to APOE4. In addition, AD neurodegeneration in specific WM tracks was shown closely correlated to cognitive decline, suggesting a potential pathologic staging marker. Our data provide novel information on the role of iron homeostasis with respective to WM degeneration, which suggests that manipulating iron homeostasis could be incorporated into the overall AD prevention and intervention strategies.
Supplemental Material
sj-docx-1-alz-10.1177_13872877251363211 - Supplemental material for Linking genetics to MRI-DTI: HFE polymorphism delays Alzheimer's disease white matter degeneration in APOE4 carriers
Supplemental material, sj-docx-1-alz-10.1177_13872877251363211 for Linking genetics to MRI-DTI: HFE polymorphism delays Alzheimer's disease white matter degeneration in APOE4 carriers by Ran Pang, Jianli Wang, Samika Kanekar, Prasanna Karunanayaka, Gela Beselia, Sangam Kanekar, Mark Meadowcroft, James R Connor, Qing X Yang and for the Alzheimer's Disease Neuroimaging Initiative in Journal of Alzheimer's Disease
Footnotes
Acknowledgments
The authors would like to thank the patients, their families, and the ADNI researchers who graciously dedicated their time and effort to make this line of research possible.
Data collection and sharing for the Alzheimer's Disease Neuroimaging Initiative (ADNI) is funded by the National Institute on Aging (National Institutes of Health Grant U19AG024904). The grantee organization is the Northern California Institute for Research and Education. In the past, ADNI has also received funding from the National Institute of Biomedical Imaging and Bioengineering, the Canadian Institutes of Health Research, and private sector contributions through the Foundation for the National Institutes of Health (FNIH) including generous contributions from the following: AbbVie, Alzheimer's Association; Alzheimer's Drug Discovery Foundation; Araclon Biotech; BioClinica, Inc.; Biogen; Bristol-Myers Squibb Company; CereSpir, Inc.; Cogstate; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; EuroImmun; F. Hoffmann-La Roche Ltd and its affiliated company Genentech, Inc.; Fujirebio; GE Healthcare; IXICO Ltd; Janssen Alzheimer Immunotherapy Research & Development, LLC.; Johnson & Johnson Pharmaceutical Research & Development LLC.; Lumosity; Lundbeck; Merck & Co., Inc.; Meso Scale Diagnostics, LLC.; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Takeda Pharmaceutical Company; and Transition Therapeutics.
Ethical considerations
This work described has been carried out in accordance with The Code of Ethics of the World Medical Association (Declaration of Helsinki) for experiments involving humans.
Consent to participate
All study participants provided informed written consent, and the study design was approved by the appropriate ethics review board.
Consent for publication
Not applicable.
Author contributions
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Institute on Aging of National Institutes Health [grant number 1R21AG064486 and R01AG070088].
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data availability statement
Data used in preparation of this article were obtained from the ADNI database. As such, the investigators within the ADNI contributed to the design and implementation of ADNI and/or provided data but did not participate in analysis or writing of this report. A complete listing of ADNI investigators can be found at: ![]() .
.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.