
Abstract
Ebola virus is a member of the Filoviridae family, which causes hemorrhagic fever in primates, exhibiting a mortality rate that can reach up to 90%. The VP35 suppresses the host IFN-β/α production by disrupting the immune responses of the host during viral infection, making it a putative target for therapeutic intervention. Herein, the UMH SuperNatural II database was mined to identify prospective VP35 inhibitors employing advanced in silico approaches. Filtration of the UMH SuperNatural II database was first conducted based on drug-likeness features. These compounds were screened towards VP35, and those exhibiting docking scores lower than 1DK, a reference ligand, further underwent molecular dynamics simulations (MDS), followed by binding energy calculations. Upon the assessed binding energy throughout 200 ns MDS, UMHSN00005544 and UMHSN00005545 disclosed superior binding affinity against VP35 compared to 1DK, with ΔGbinding values of −35.5, −34.9, and −29.3 kcal/mol, respectively. Energetic and structural evaluations were conducted for the identified natural compounds in complex with VP35 over 200 ns MDS. Post-MD analyses demonstrated the significant constancy of the investigated complexes. RMSD values averaged 0.14, 0.13, and 0.12 nm for UMHSN00005544, UMHSN00005545, and 1DK bound to VP35 over 200 ns MDS, indicating stable protein-ligand conformations. Furthermore, the ADMET characteristics of the identified natural compounds were assessed, revealing favorable pharmacokinetic and non-toxicity profiles. Density functional theory computations unveiled the electronic stability and chemical reactivity of the identified natural compounds. The obtained outcomes affirmed the substantial therapeutic potential of UMHSN00005544 and UMHSN00005545 as prospective candidates for combating EBOV, thereby necessitating further experimental investigations.
Keywords
Introduction
Ebola virus (EBOV) is an enveloped, negative-sense RNA virus that belongs to the Filoviridae family.1,2 It causes intense hemorrhagic fevers and acute systemic illnesses, collectively referred to as Ebola virus disease (EVD). 3 EVD is one of the most severe hemorrhagic fevers affecting humans and nonhuman primates, with recorded fatality rates reaching as high as 90% in certain outbreaks. 4 A West African outbreak from 2013 to 2016 showed a significant epidemic potential of the EBOV, which resulted in over 28,000 reported cases and approximately 11,000 deaths. 5 As an emerging zoonotic disease, EVD is primarily spread by fruit bats from the Pteropodidae family. 6 EVD typically begins with vague, flu-like manifestations before escalating into a pronounced cytokine storm, massive fluid loss, and dehydration. 7 This progression can lead to critical complications, including hemorrhage, septic shock, systemic capillary leak syndrome, and multi-organ failure, ultimately resulting in mortality. 7 The pathology of EBOV remains unclear despite dedicated research initiatives. There is still a substantial need for extensive prevention strategies and advances in prophylactic and therapeutic interventions, according to a better comprehension of the mechanisms that govern the host-pathogen relationship. 8
As part of the EBOV genome, seven structural proteins are encoded, including the glycoprotein (GP), the nucleoprotein (NP), the viral proteins (VP30, VP35, VP24), and the RNA-dependent RNA polymerase (L).9–12 Additionally, it encodes non-structural/small secretory glycoprotein (sGP/ssGP). EBOV viral polymerase 35 (VP35) suppresses the host’s immunological response, including the production of interferon (IFN) stimulated by sensory receptors, and is considered a viral assembly factor.13–17 Because of its dual and central function in immune suppression and viral replication, VP35 represents a promising target. By inhibiting VP35, EBOV amplification can be controlled, thereby protecting humans from potentially fatal infections.18–20 However, to date, no medication has received approval as a VP35 inhibitor. Consequently, there remains a necessity to investigate the potentialities of alternative active pharmaceutical compounds targeting VP35.
Natural products (NPs) are considered a crucial source of pharmaceutical medications. Numerous widely used antiviral treatments have been derived from natural origins, such as acyclovir, cidofovir, and famciclovir.21–23 A recent study has highlighted the potential of natural compounds as effective inhibitors of viral proteins, including VP35, using computational approaches.
24
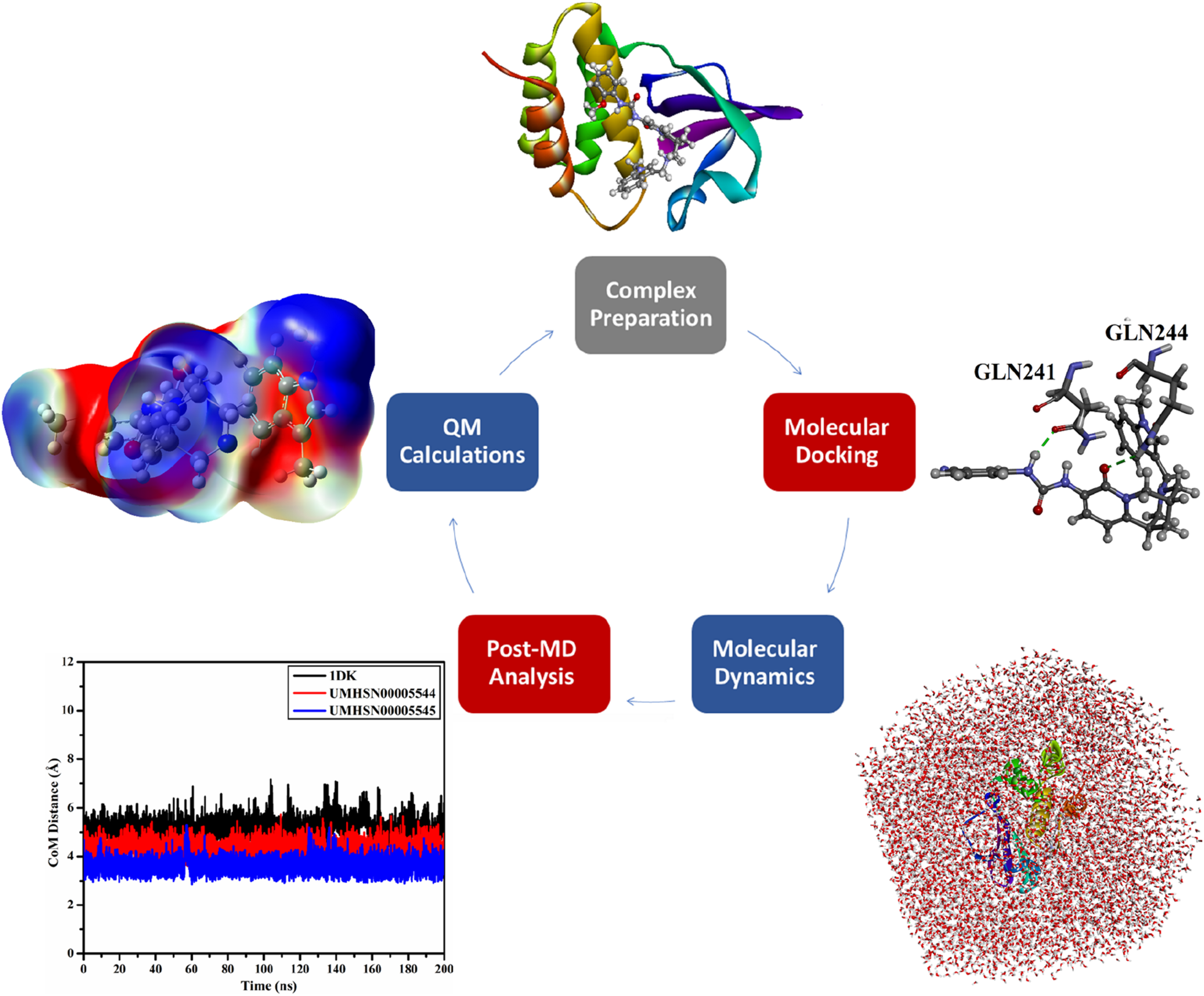
In the current study, the UMH SuperNatural II database, encompassing >326,000 compounds, was first filtered according to its drug-likeness features (i.e., Ro5). The obtained compounds from Ro5-based filtration were subsequently submitted to docking computations towards VP35 to investigate their binding modes and docking scores. Upon the predicted docking scores, the most potent natural compounds in complex with VP35 were advanced for molecular dynamics simulations (MDS) over 200 ns. Additionally, the binding energy over MDS was evaluated employing the MM-GBSA approach. Moreover, the energetic and structural analyses of the identified natural compounds bound to VP35 were inspected. As well, the pharmacokinetics and toxicity of the identified natural compounds were predicted. Furthermore, quantum mechanical computations were employed to gain a deeper comprehension of the electronic and geometric properties of the identified natural compounds. Figure 1 depicts a schematic illustration of the computational methods utilized for filtrating the UMH SuperNatural II database as prospective inhibitors for VP35. The current results unveiled that the identified natural compounds were promising to inhibit the VP35 activity and act as anti-EBOV drug candidates. Schematic representation of the in silico methods utilized in UMH SuperNatural II database filtration as prospective inhibitors for VP35.
Computational methods
VP35 preparation
The three-dimensional coordinates of the VP35 bound to 1DK (PDB entry: 4IBB; Resolution: 1.75 Å 25 ) were downloaded from the RCSB Protein Data Bank. 26 All heteroatoms and water molecules were discarded for preparation purposes. The ionization states of titratable amino acids were checked through the H++ server. 27 Besides, all missing hydrogens were added.
Database preparation
The UMH SuperNatural II database, encompassing >326,000 compounds, was obtained in SDF format from Prof. Encinar’s website (https://docking.umh.es/downloaddb). Furthermore, to ensure the integrity and uniqueness of the database, any duplicate compounds identified by an identical International Chemical Identifier (InChIKey) were removed. 28 The 3D structure for each compound was subsequently generated utilizing Omega2 software.29,30 MMFF94S within the SZYBKI software was used for executing an energetical minimization for all generated structures.31,32 The protonation states of the natural compounds were determined using the Fixpka algorithm inside the QUACPAC software. 33 The prepared database can be found at https://www.compchem.net/ccdb.
Drug-likeness properties
UMH SuperNatural II compounds were filtered based on Lipinski’s rule of five (Ro5).34,35 The following criteria were used for filtering: the number of hydrogen bond acceptors/donors (HBA/HBD) should not exceed 10/5, respectively. Additionally, the molecular weight (MW) should not be more than 500 Da. The partition coefficient (LogP) value should be <5.0. Moreover, the topological polar surface area (TPSA) of a molecule, which influences its capacity to permeate cell membranes, should be ≤140 Å2. Only 128,126 natural compounds obeyed Ro5 criteria. Therefore, only these compounds were subjected to a virtual screening against VP35.
Docking computations
Herein, all docking predictions were accomplished employing the AutoDock Vina (version 1.1.2) software. 36 The VP35 structure was converted into the PDBQT format using MGLTools. 37 Two levels of docking precision were conducted, namely, standard and expensive docking computations. All docking parameters, with the exception of the exhaustiveness number, were configured to their default settings. For the docking predictions, the exhaustiveness parameter was set to 10 for standard docking computations, which is sufficient for initial screening of an extensive database. For expensive docking computations, an exhaustiveness value of 200 was utilized, which significantly increases the search thoroughness and improves the chances of identifying the global minimum binding conformation.36,38 The docking grid was defined at 15 Å × 15 Å×15 Å. A grid spacing of 1.0 Å was used for the docking estimations. The center of the grid was stationed at 2.022, 21.568, and 2.037 (in XYZ coordinates). AutoDock Vina1.1.2 software utilizes a hybrid scoring function that integrates knowledge-based and empirical terms, responsible for H-bonding, hydrophobic interactions, steric complementarity, and conformational entropy to evaluate ligand docking score. 36
Molecular dynamics simulations
All MDS were accomplished utilizing AMBER software (version 20). 39 Detailed information regarding the implemented MD protocol is elaborated elsewhere.40–43 The investigated natural compounds were characterized by the General AMBER Force Field (GAFF2). 44 Meanwhile, the VP35 was parametrized with the AMBER force field 14SB. 45 Herein, both implicit and explicit solvent MDS were executed.
In the implicit solvent MDS, the atomic charges of the investigated natural compounds were assigned using the AM1-BCC method. 46 Neither periodic boundary nor cutoff conditions were applied to nonbonded interactions. Additionally, the solvation impact was assessed employing the generalized Born model (igb = 2). Furthermore, the inspected complexes were minimized for 500 cycles and subsequently heated to 310 K for 10 ps. The ligand-VP35 complexes were then equilibrated for 50 ps. Subsequently, the production phase for the ligand-VP35 complexes was accomplished through 1 ns MDS.
In explicit solvent MDS, the inspected natural compounds were optimized at the HF/6-31G* level with the help of Gaussian09 software. 47 The atomic charges of the optimized compounds were determined through the restrained electrostatic potential (RESP) approach at the same level of theory utilized for geometric optimization. 48 The investigated ligand-VP35 complexes were solvated by the TIP3P model in an octahedron water box with a box dimension of 12 Å. Proper Na+/Cl− ions were subsequently added to neutralize all investigated complexes. The ionic strength of the solvated complexes was maintained at 0.15 M NaCl. The final simulation box volume, recorded after solvation and ions addition, was about 261.4 nm3 for the investigated complexes. Moreover, the investigated natural compounds complexed with VP35 were minimized through 5000 cycles and annealed to 310 K for 50 ps. Following the heating phase, the complexes were subjected to equilibration for 10 ns to ensure that the investigated complexes reached a stable state before running the production phase. The production phases were performed over 10 and 200 ns MDS. For post-MD analyses, snapshots were captured every 10 ps. All MDS were executed utilizing the CUDA-accelerated version of the pmemd module included in the AMBER20 suite. All visualizations were fulfilled using Discovery Studio Visualizer. 49
Binding energy computations
The binding energies (ΔGbinding) of the identified natural compounds bound to VP35 were computed utilizing the molecular mechanical-generalized Born surface area (MM-GBSA) approach.
50
More exactly, the MMPBSA. py module was employed to execute binding energy computations on snapshots extracted every 10 ps over a simulation time.
51
The ΔGbinding was calculated in accordance with the subsequent equation:


EvdW stands for the van der Waals energy. Eele and GGB refer to electrostatic and general-born solvation energies, respectively. GSA is the surface area energy. Entropy contribution was neglected due to its high computation cost.52,53
Principal component analysis (PCA)
PCA was performed to assess the covariance of atomic movements and the dynamic behavior of protein loops. 54 Trajectory processing was carried out using the PTRAJ module inside the AMBER20 software package, where water molecules and ions were eliminated. The collected trajectories over the MD course were then aligned with their respective fully optimized conformations to eliminate any overall translational and rotational motions. Covariance matrices of the C α atomic fluctuations were then generated, and the first two principal components (PC1 and PC2) were calculated. PCA was performed on 20,000 trajectories for each C α atom in both the apo-VP35 and ligand-bound systems. The first two eigenvectors of the covariance matrices corresponded to the extracted PC1 and PC2.
ADMET properties
The ADMET properties involved absorption, distribution, metabolism, excretion, and toxicity. Therefore, pharmacokinetics for the identified natural compounds were estimated via the pkCSM online server. 55 Absorption (A) was predicted by P-glycoprotein substrate, skin permeability, and human intestinal absorption (HIA). Moreover, the capacity of the identified natural compounds to act as inhibitors for P-glycoprotein (P-gp) type I and type II was also anticipated. Distribution (D) was predicted according to the central nervous system (CNS) and blood-brain barrier (BBB) permeabilities. Metabolism (M) was predicted in accordance with CYP2D6 and CYP3A4 substrates and inhibitors. Moreover, excretion (E) was predicted via renal OCT2 substrate. Finally, the toxicity (T) was expected according to the AMES toxicity, hERG I inhibitor, and skin sensitization toxicity. 56
DFT calculation
The final trajectory of the identified natural compounds derived from MDS over a duration of 200 ns was retrieved and geometrically optimized at the M062X/6-311+G** level utilizing Gaussian09 software.
47
The electrostatic potential (ESP) analysis was then conducted for the optimized inhibitors.
57
Within the framework of ESP analysis, molecular electrostatic potential (MEP) maps were evaluated and visualized on an electron density isosurface set at 0.002 au. A thorough comprehension of the electronic characteristics of the identified natural compounds was achieved through the implementation of the frontier molecular orbitals (FMOs) theory. The lowest unoccupied and highest occupied molecular orbitals (LUMO/HOMO) distributions were plotted for the optimized structures of the identified natural compounds. In a similar quantitative approach, HOMO/LUMO energy levels (EHOMO/ELUMO) were assessed. According to the acquired orbital energies, the energy gap (Egap) and Fermi level (EFL) values were evaluated employing equations (3) and (4), respectively.


Furthermore, the ionization potential (IP) and electron affinity (EA) were calculated using the ELUMO and EHOMO, as outlined in equations (5) and (6), respectively.


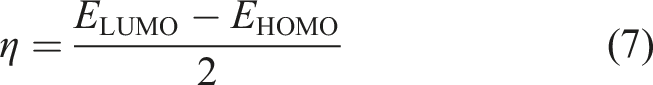

Results and discussions
Docking protocol validation
The docking protocol employed in this study was validated using AutoDock Vina1.1.2 software, with results compared with available experimental data to ensure its reliability. To assess the accuracy of the docking protocol, the co-crystallized 1DK ligand was re-docked into the VP35 active site. The overlapping between the original binding pose and the docked mode is shown in Figure 2. As shown in Figure 2, the matching between the original binding pose and the predicted docked mode of the 1DK was noticed with an RMSD value of 0.63 Å. Generally, this observed RMSD value was below 2.0 Å, indicating reliability in anticipating the correct binding mode.59,60 As well, the estimated docking score of 1DK with VP35 was −6.7 kcal/mol. Analyzing 1DK docking mode with the VP35 active site displayed that the CO group of thiophene-2-carbaldehyde displayed an H-bond with the NH2 of GLN241 (1.55 Å). IDK also interacted with PHE328 by pi-sulfur interaction and exhibited pi-sigma interaction with LYS248. In accordance with the current results, the validated protocol was applied to hunt the most potent inhibitors against VP35 from the UMH SuperNatural II database. (a) Overlapping between the original binding pose (mauve) and the predicted docked mode (green) of 1DK, (b) three-dimensional and (c) two-dimensional molecular interactions of the predicted docking mode of the 1DK inhibitor within the VP35 active site.
Database mining
The estimated docking scores, conventional H-bonds, and two-dimensional chemical structures of 1DK and the top two natural compounds towards VP35 a .

aData organized based on the expensive docking score.
UMHSN00005545 exhibited the most favorable docking of −9.4 kcal/mol. A detailed investigation of the docking mode of UMHSN00005545 inside the VP35 active site manifested that the nitrogen-hydrogen (NH) moiety of the N-(2-methoxyphenyl) formamide fragment exhibited an H-bond with the CO of the GLN244 (2.22 Å) (Figure 3). In addition, the carbonyl of the 3-aminopyridin-2(1H)-one group established an H-bond with the NH2 of the GLN241 (2.43 Å). Two- and three-dimensional binding patterns of (a) UMHSN00005545 and (b) UMHSN00005544 within the VP35 active site.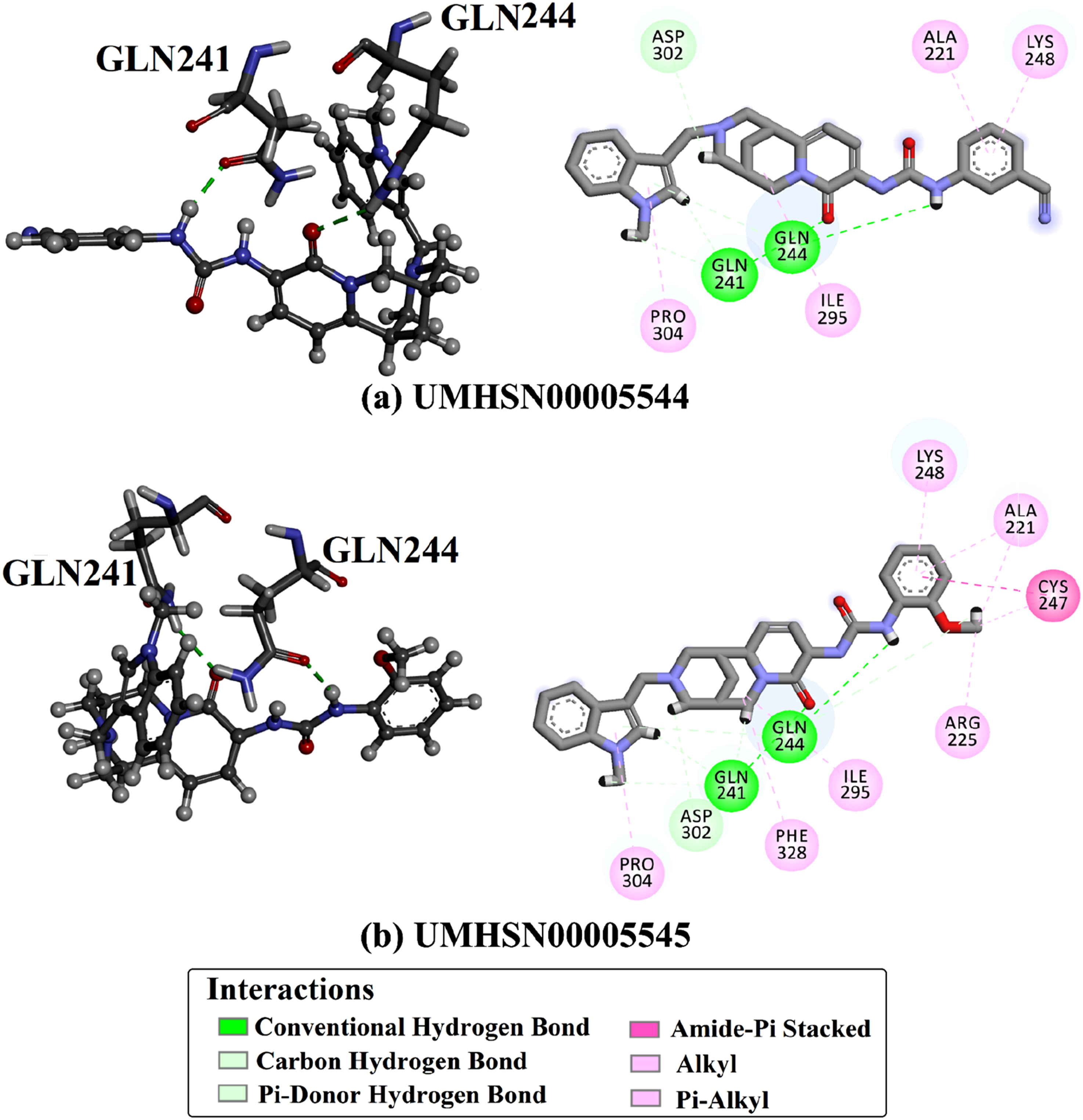
UMHSN00005544 achieved the second-lowest docking score, recorded at −9.2 kcal/mol. Analyzing the UMHSN00005544 docking mode bound to VP35 disclosed that the NH of the 3-(methylamino) benzonitrile moiety formed an H-bond with the CO of the GLN244 (2.05 Å) (Figure 3). Additionally, the CO of the 1,6-dimethyl-3-(methylamino) pyridine-2(1H)-one moiety demonstrated an H-bond with the NH2 of the GLN241 (2.54 Å).
As apparent in Figure 3, the analysis of interactions further disclosed the presence of different types of bonds, including pi-cation, pi-sigma, carbon H-bonds, and amide-pi stacked interactions between UMHSN00005545 and UMHSN00005545 with the proximal residues of the VP35 active site. Of note, the observed interactions of the identified natural compounds with adjacent residues were almost identical, indicating a similarity in the binding modes of these compounds within the VP35 active site.
MDS
Binding energies for 1DK-, UMHSN00005544-, and UMHSN00005545-VP35 complexes over 10 and 200 ns MDS.

Binding energies decomposition of 1DK-, UMHSN00005544-, and UMHSN00005545-VP35 complexes through 200 ns MDS.

To accurately identify the residues within the VP35 active site that interact with the selected natural compounds throughout 200 ns MDS, a detailed per-residue decomposition was performed. Figure 4 illustrates the per-residue decomposition for only residues exhibiting ΔGbinding < −0.5 kcal/mol. The findings, as represented in Figure 4, revealed that the GLN244 plays a pivotal role, displaying the most substantial contribution to ΔGbinding with average values of −3.9, −4.5, and −0.7 kcal/mol for UMHSN00005544, UMHSN00005545, and 1DK, respectively. The energy participations of the fundamental residues in the binding of 1DK, UMHSN00005544, and UMHSN00005545 with VP35 throughout 200 ns MDS.
Additionally, the GLN241 was observed to also provide a significant contribution to the ΔGbinding, with mean values of −3.7, −2.1, and −3.1 kcal/mol for UMHSN00005544, UMHSN00005545, and 1DK, respectively (Figure 4). Significantly, these findings corresponded with the anticipated docking modes of the identified natural compounds and 1DK inside the VP35 active site (Figure 3).
Energetic and structural analyses
To thoroughly assess the constancy of the UMHSN00005544, UMHSN00005545, and 1DK bound to the VP35 throughout the 200 MDS, several energetic and structural evaluations were executed.
Binding affinity per-trajectory
To evaluate the energetic constancy of the UMHSN00005544-, UMHSN00005545-, and 1DK-VP35 complexes over 200 ns MDS, the correlation between binding energy and time was assessed (Figure 5(a)). Figure 5(a) demonstrates the remarkable stability observed throughout the 200 ns MDS for the UMHSN00005544-, UMHSN00005545-, and 1DK-VP35 complexes, with mean ΔGbinding values of −35.5, −34.9, and −29.3 kcal/mol, respectively. These outcomes verified the overall steadiness of the identified natural compounds bound to VP35 over the 200 ns MDS. (a) Estimated binding affinities, (b) CoM distances, and (c) RMSD of 1DK (black), UMHSN00005544 (red), and UMHSN00005545 (blue) bound to VP35 throughout 200 ns MDS.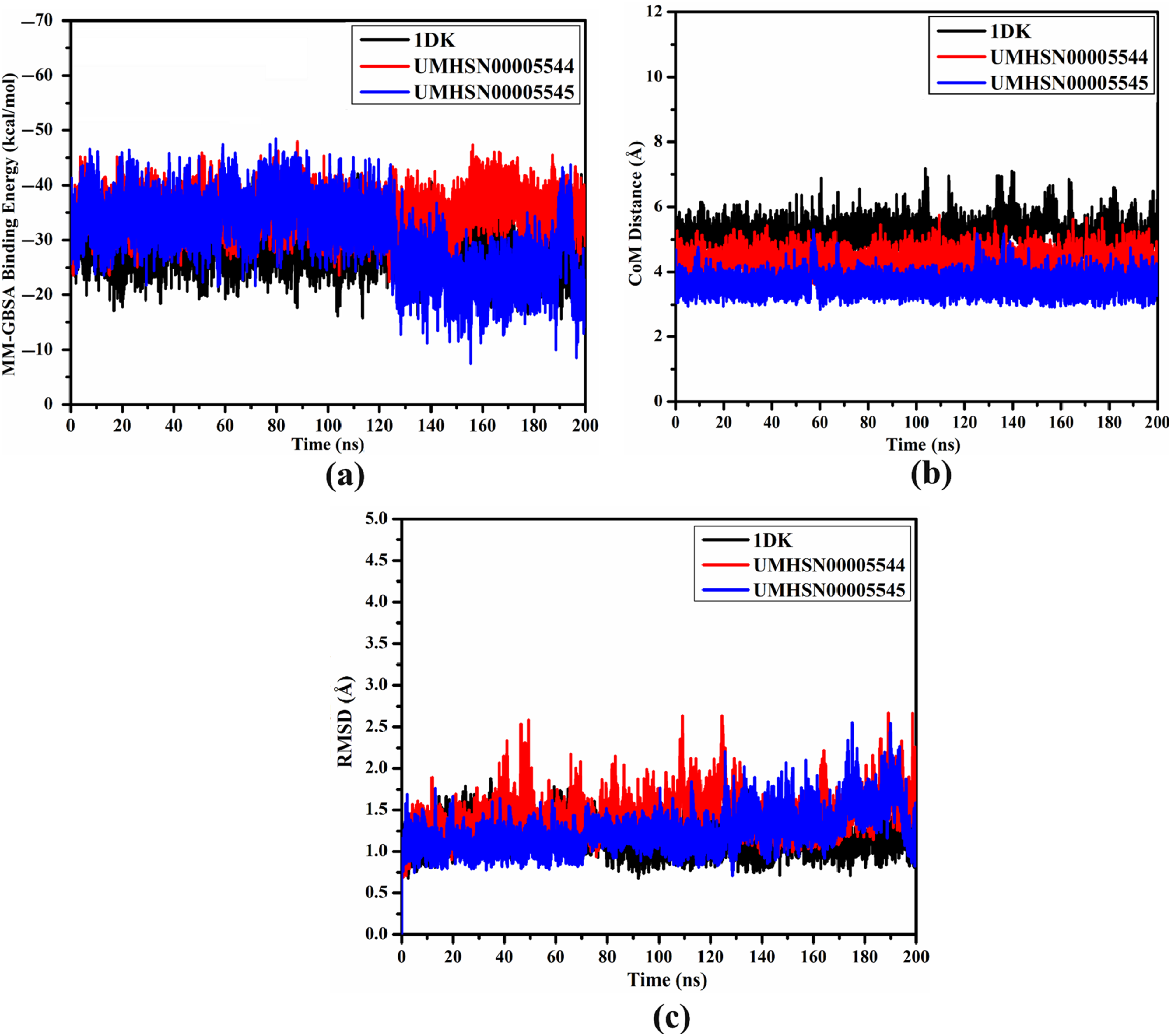
Distance of center-of-mass (CoM)
In order to verify the steadiness of UMHSN00005544, UMHSN00005545, and 1DK in complex with VP35, the distance of CoM between the identified natural compounds and GLN244 was measured (Figure 5(b)). As depicted in Figure 5(b), the high stabilization of UMHSN00005544, UMHSN00005545, and 1DK complexed with VP35 was observed with mean CoM distances of 4.3, 4.5, and 4.2 Å, respectively. The current data proved the great constancy of all investigated complexes over 200 ns MDS.
Root-mean-square deviation (RMSD)
The RMSD of the backbone atoms of the entire complex was gauged to monitor the conformational variation of UMHSN00005544, UMHSN00005545, and 1DK complexed with VP35 throughout 200 ns MDS (Figure 5(c)). The inspected complexes conserved their constancy till the termination of the 200 ns MDS (Figure 5(c)). The evaluated RMSD values were 1.4, 1.3, and 1.2 Å for UMHSN00005544, UMHSN00005545, and 1DK bound to VP35 over the simulation time. The present results proved that UMHSN00005544, UMHSN00005545, and 1DK were tightly bound and maintained the structural integrity.
H-bond analysis
The constancy of UMHSN00005544, UMHSN00005545, and IDK complexed with VP35 was estimated by measuring the H-bond number between the identified natural products and the proximal residues of the VP35 active site over the 200 ns MDS (Figure 6). The average H-bond numbers were 2, 2, and 2 for UMHSN00005544-, UMHSN00005545-, and IDK-VP35 complexes (Figure 6). Generally, these results suggested a strong alignment with the anticipated docking depicted in Figure 3. Additionally, the H-bond assessment provided support for the constancy of the identified natural compounds within the VP35 active site throughout 200 ns MDS. H-bond number for (a) 1DK, (b) UMHSN00005544, and (c) UMHSN00005545 bound to VP35 over 200 ns MDS.
Root-mean-square fluctuation (RMSF)
RMSF is a parameter that is frequently employed to evaluate the flexibility of each residue during MDS. RMSF illustrates the fluctuations in the positions of residues from the initial to the final trajectories of the MDS, reflecting the deviations in the structural configurations of the target.
64
To comprehend the impact of structural variations on the target’s elasticity, the RMSF was computed over 200 ns MDS (Figure 7(a)). Higher RMSF values indicate greater elasticity of the receptor throughout the MDS. The average RMSF values were found to be 0.115, 0.141, 0.175, and 0.138 nm for the apo-, UMHSN00005544-, UMHSN00005545-, and 1DK-VP35 complexes, respectively (Figure 7(a)). These results suggested that the receptor flexibility is affected by its inhibitor interactions, with the identified natural compounds promoting a more dynamic behavior in VP35, potentially affecting its functional role in various biological processes. (a) RMSF, (b) Rg, and (c) SASA of apo-VP35 (cyan), 1DK-VP35 (black), UMHSN00005544-VP35 (red), and UMHSN00005545-VP35 (blue) throughout 200 ns MDS.
Radius of gyration (Rg)
The Rg is an important parameter used to evaluate the equilibrium configurations of the entire system, providing insight into the compactness of the receptor during MDS. Protein compactness can vary during receptor-ligand interactions. 65 To examine how residue flexibility impacts protein compactness, the Rg was calculated for the VP35 complexed with UMHSN00005544, UMHSN00005545, and 1DK throughout 200 ns MDS (Figure 7(b)). The average Rg values were 1.47, 1.49, 1.45, and 1.44 nm for the apo-, UMHSN00005544-, UMHSN00005545-, and 1DK-VP35 complexes, respectively. These findings indicated that the VP35 structure became more compact and stable after complexation with UMHSN00005544 and UMHSN00005545.
Solvent-accessible surface area (SASA)
Water plays a critical role in determining the stability, structure, function, and dynamics of proteins. SASA is a useful indicator for measuring the extent to which protein residues are accessible to water molecules. 58 Figure 7(c) shows the SASA of the VP35 upon binding with UMHSN00005544, UMHSN00005545, and 1DK over the course of 200 ns MDS. The SASA plots revealed similar trends for UMHSN00005544-, UMHSN00005545-, and 1DK-VP35 complexes. Compared to apo-VP35, the SASA of VP35 slightly decreased upon binding with 1DK (Figure 7(c)). In contrast, when UMHSN00005544 and UMHSN00005545 were bound to VP35, the SASA value slightly increased. The average SASA values were 62.2, 63.2, and 62.6, 61.6 nm2 for the apo-, UMHSN00005544-, UMHSN00005545-, and 1DK-VP35, respectively. These results implied that the identified natural compounds and 1DK unveiled minimal influence on the solvent accessibility of VP35.
PCA analysis
PCA was used to inspect the structural dynamics of both the apo-, UMHSN00005544-, UMHSN00005545-, and IDK-VP35 over 200 ns MDS. To get a deeper understanding of the conformational changes over the simulations, a PCA-based clustering approach was utilized to group conformations based on their structural similarities.
66
Figure 8 displays the distribution of motions along the first two principal components (PC1 and PC2) for apo-VP35 and UMHSN00005544-, UMHSN00005545-, and IDK-VP35 complexes. The displayed scatterplot illustrates the projection of simulation frames onto the PC1 and PC2 axes, revealing distinct configurational sampling between the two systems. The apo-VP35 system demonstrated a broader distribution and lower correlation of movements, indicating greater flexibility of the residues. These outcomes evinced that the binding of the identified natural compounds induced considerable changes in the dynamics of VP35. PCA of MDS trajectories for apo-VP35 and (a) 1DK-VP35, (b) UMHSN00005544-VP35, and (c) UMHSN00005545-VP35.
ADMET analysis
ADMET features of UMHSN00005544, UMHSN00005545, and 1DK as prospective ant-EBOV drug candidates.

DFT calculation
MEP maps were generated for the optimized structures retrived from the last trajectory of the 200 ns MDS for UMHSN00005544, UMHSN00005545, and 1DK (Figure 9). From the pertinent MEP maps, red areas were detected on the O and N atoms of the identified natural compounds and 1DK, indicating their nucleophilic nature. Additionally, clear blue regions were observed surrounding the C and H atoms of the identified natural compounds, indicating their electrophilic characteristics. By examining the MEP maps of the compounds under investigation, it was found that these compounds possess the capability of establishing H-bonds with the essential residues located within the VP35 active site. Generated MEP maps for the optimized (a) UMHSN00005544, (b) UMHSN00005545, and (c) 1DK.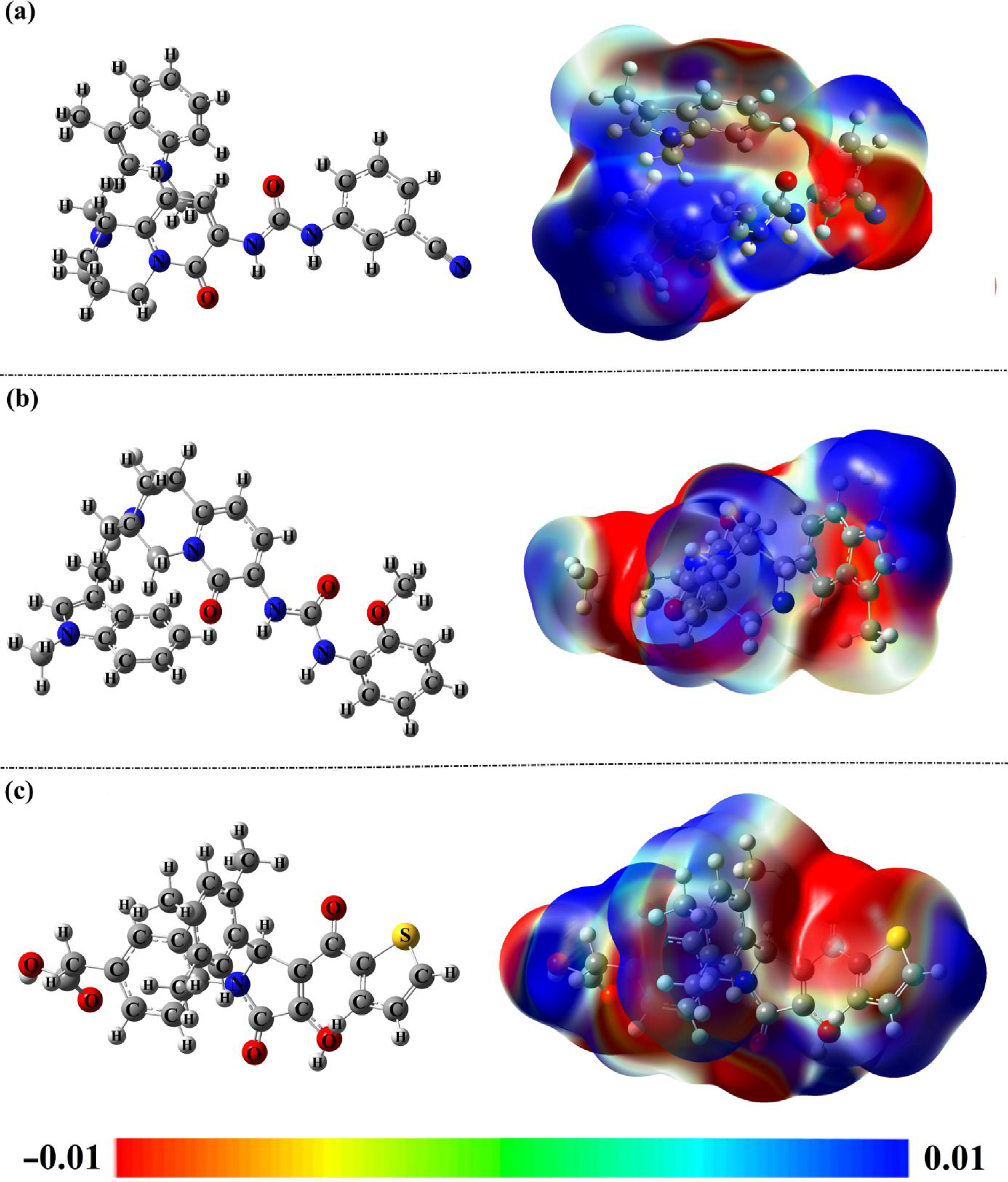
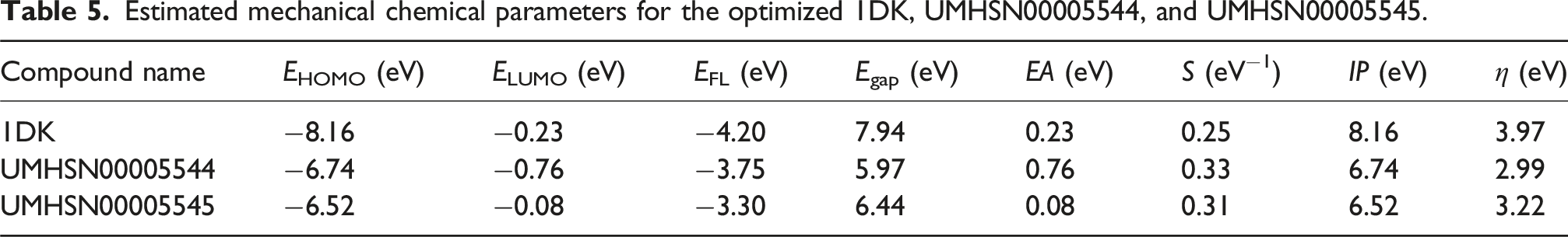
Estimated mechanical chemical parameters for the optimized 1DK, UMHSN00005544, and UMHSN00005545.

Distribution of HOMO and LUMO of the optimized (a) UMHSN00005544, (b) UMHSN00005545, and (c) 1DK.
Of note, the HOMO distribution, as depicted in Figure 10, was mainly observed around the N and O atoms, while the LUMO distribution was found to be associated with the H atoms of the identified natural compounds and 1DK.
Regarding the data in Table 5, EHOMO/ELUMO values were −6.74/–0.76, −6.52/–0.08, and −8.16/–0.23 eV for UMHSN00005544, UMHSN00005545, and 1DK, respectively. Moreover, UMHSN00005544, UMHSN00005545, and 1DK exhibited low Egap with values of 5.97, 6.44, and 7.94 eV, respectively, signifying their considerable levels of chemical reactivity. According to their Egap values, the identified compounds seem to follow this sequence of chemical reactivity: UMHSN00005544 > UMHSN00005545 > 1DK. In a similar manner, the EFL values were found to be −3.75, −3.30, and −4.20 eV for UMHSN00005544, UMHSN00005545, and 1DK, respectively.
The evaluation of global indices of reactivity seeks to distinctly clarify the chemical properties of the investigated compounds. For the identified natural compounds, a set of global indices of reactivity comprising EA, IP, S, and η was computed (Table 5).
The electronic stability and chemical reactivity of the identified natural compounds can be assessed using their hardness (η) values. The η of the identified natural compounds and 1DK increased in the following order: UMHSN00005544 < UMHSN00005545 < 1DK. As well, UMHSN00005544, UMHSN00005545, and 1DK demonstrated promising IP with values of 6.74, 6.52, and 8.16 eV, respectively. The degree of S for the identified natural compounds and 1DK decreased in the subsequent order: UMHSN00005544 > UMHSN00005545 > 1DK (Table 5).
Conclusion
The Ebola virus represents one of the most lethal infectious diseases to affect humans. VP35 plays a vital function in obstructing the host’s interferon-alpha/beta (IFN-α/β) production, making it an appealing target for finding novel drugs that can fight EBOV infection. In this study, the UMH SuperNatural II database, encompassing >326,000 compounds, was mined to identify promising VP35 inhibitors utilizing advanced in silico approaches. Upon the docking scores, the most promising natural compounds with docking scores of less than 1DK (reference inhibitor, calc. −6.7 kcal/mol) were advanced for MDS, accompanied by the calculation of MM-GBSA binding energy. Upon MM-GBSA//200 ns MDS, UMHSN00005544 and UMHSN00005545 exhibited higher binding affinities than 1DK with ΔGbinding values of −35.5, −34.9, and −29.3 kcal/mol, respectively. Post-MD analyses underscored the stability of the identified natural compounds bound to VP35 throughout the 200 ns MDS. UMHSN00005544 and UMHSN00005545 also revealed favorable pharmacokinetic properties, as indicated by their ADMET profiles. According to FMO calculations, the Egap values for UMHSN00005544, UMHSN00005545, and 1DK were 5.97, 6.44, and 7.94 eV, respectively, suggesting that the identified natural compounds are stable and inherently bioactive. Given the promising potential of these natural compounds, further exploration through detailed experimental studies is warranted to facilitate the development of innovative therapeutics aimed at combating EBOV infections.
Supplemental Material
Supplemental Material - Harnessing SuperNatural database to identify VP35 inhibitors as anti-Ebola drug candidates: A multistage In silico study
Supplemental Material for Harnessing SuperNatural database to identify VP35 inhibitors as anti-Ebola drug candidates: A multistage In silico study by Abeer A. M. Hasb, Gamal A. H. Mekhemer, Peter A. Sidhom, Ahmed Rady, Yanshuo Han, and Mahmoud A. A. Ibrahim in Antiviral Therapy
Footnotes
Acknowledgements
The authors extend their appreciation to the Ongoing Research Funding Program, (ORF-2026-691), King Saud University, Riyadh, Saudi Arabia, for funding this work. The computational work was completed with resources provided by the CompChem Lab (Minia University, Egypt, hpc.compchem.net), Center for High-Performance Computing (Cape Town, South Africa, https://www.chpc.ac.za/), and Bibliotheca Alexandrina (![]() ).
).
Author contributions
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data Availability Statement
Data included in article/supp. material/referenced in the article.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.