
Abstract

Poster No. 60
Abstract ID No. 1494785
Venous inflammation might be one of the features of VEXAS syndrome and associated thrombosis
Hazan Karadeniz1, Mahinur Cerit1, Seyma Yildiz1, Abdurrahman Tufan2, Yogen Kanthi, MD3 1Gazi University Faculty of Medicine, 2National Human Genome Research Institute/NIH, 3National Institutes of Health
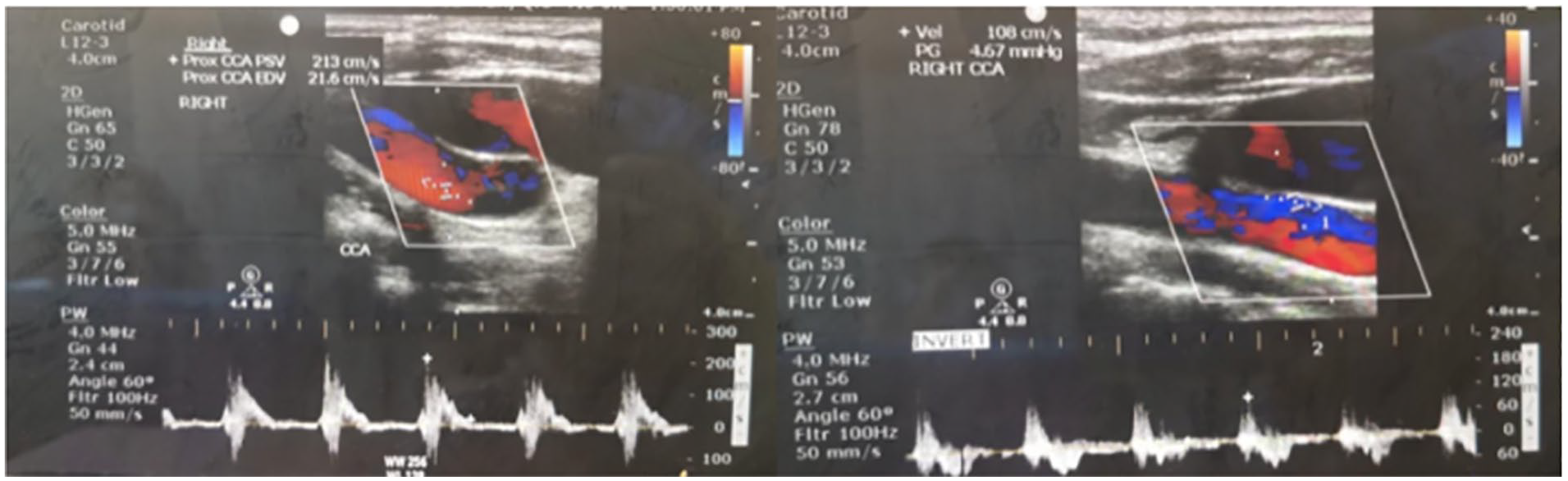
Background: VEXAS syndrome is an often fatal, X-linked disease characterized by hematological dysplasia overlapping with severe autoinflammatory manifestations. Thrombosis, predominantly as unprovoked venous thrombosis, has been reported in up to 56% of VEXAS patients. The exact pathogenesis of thrombosis is yet to be elucidated. Chronic inflammation, vasculitis, endothelial dysfunction, disrupted anticoagulant mechanisms, or prothrombotic antibodies are suggested mechanisms. The clinical and pathogenetic similarities with BS, we thought venulitis might be the underlying cause of thrombosis in VEXAS syndrome. Herein, we report venulitis in three consecutive VEXAS patients with the demonstration of severe thickening of the walls of large veins.
Case presentation: Bilateral lower extremity common femoral veins (CFV) were examined with Doppler ultrasound in craniocaudal direction for the presence of thrombi and venous insufficiency. Beside features of CFV, venous wall thickness (VWT) of the internal jugular veins (IJV) in patients and healthy control were assessed. All measurements were performed thrice, and the arithmetic mean was given as the result. VWT measurements previously shown in Behçet’s patients were taken as reference in our study. VEXAS patients had thickened walls on ultrasound in all examined large veins. Patients with extensive thrombosis (Case#1 and Case #3) had the most severe VWT of CFV on the affected side, while the latter case (Case#2) had equivalent VWT in both CFVs. Also, we determined increased IJV thickness in both VEXAS (Case#1 and Case#2) patients compared to healthy controls.
Conclusions: Consequently, our findings suggest that venulitis might be one of the features of VEXAS syndrome and the underlying mechanism for thrombosis.

Poster No. 61
Abstract ID No. 1511177
IGG4-Related disease manifestations post-COVID-19 infection: a consequence or a coincidence?
Hamdan AlAwadhi, MD, Yosef Manla, MD, Firas Al Badarin, MD, Batool Abuhalimeh, MD, Houssam Younes, MD Cleveland Clinic Abu Dhabi
Background: Long COVID is yet to be fully understood. Limited data are available on the manifestations of IgG4-related diseases (IgG4-RD), including aortitis and retroperitoneal fibrosis in those who recovered from COVID-19 infection.
Case presentation: A previously healthy 39-year-old male with a history of symptomatic COVID-19 ten months earlier presented to our center with a 1-day history of worsening abdominal pain and vomiting. The patient reported that the pain started four months ago. His laboratory findings were significant for high inflammatory markers, a CRP of 25mg/l, and ESR>119 mm/hr despite a normal white blood cell count. CT scan of the abdomen and pelvis revealed a retroperitoneal mass encasing the infrarenal aorta, the bilateral common iliac arteries, and the right ureter causing proximal mild-to-moderate right hydroureteronephrosis. The patient underwent abdominal and pelvic MRA, which showed a T2 signal intensity mass-like lesion encasing the aorta. Abdominal aorta doppler ultrasound showed mid to distal lesions involving the tunica media and adventitia. Further laboratory tests with flow cytometry, complement, serum protein electrophoresis, infectious work-up including HIV screening, and rheumatological panels were all negative except for an elevation in the IgG-4 1.23 g/dl. The patient PET scan was consistent with active retroperitoneal fibrosis. However, since the criteria for the diagnosis of IgG-4 RD has not been met, the patient’s symptoms were attributed to long COVID. The patient was started on a prednisolone regimen and was followed up two months later with a CT scan of the abdomen and pelvis, which showed improvement in his retroperitoneal fibrosis.
Conclusions: Our patient likely had post-COVID-19 infection aortitis and peri-aortic fibrosis as he developed his symptoms eight months after the infection. It is postulated that the virus over-activates the cellular and humoral immunity systems, which might have resulted in these long-term sequelae. In patients with peri-aortic fibrosis, a history of COVID-19 infection can help guide the diagnosis and management to avoid fetal complications.
Poster No. 62
Abstract ID No. 1511239
Acrocyanosis and pernio after an arctic blast
Merry Ellen D. Barnett, MD, Aditya M. Sharma, MBBS, Randy K. Ramcharitar, MD
University of Virginia
Background: Acrocyanosis is a functional vascular disorder characterized by blue digital discoloration and pernio is an inflammatory cutaneous condition resulting in digital erythema and local edema. Both can be triggered by cold exposure, but acrocyanosis tends to be persistent while pernio is transient. We describe a case of acrocyanosis evaluated at our institution to review the clinical presentation and highlight characteristics that can be helpful in diagnosis.
Case presentation: A 78-year-old man presented to clinic for evaluation of persistent discoloration of the first and second digits on bilateral feet. Past medical history was significant for hypertension, hyperlipidemia, and 20 pack-year smoking history (quit five years ago). Symptoms began approximately two months prior to presentation. A few days after an arctic blast, he developed erythematous, slightly swollen, and tender to touch macules on the distal aspect of bilateral dorsal toes. These symptoms had resolved within two weeks of him making a dedicated effort to wear shoes and socks, but he was left with persistent, painless, blue discoloration of the first and second toes bilaterally. He also had a documented 9 kg weight loss in the past 9 months. Physical exam revealed normal distal pulses and blue discoloration of bilateral toes. Noninvasive vascular testing was normal apart from mildly dampened digital photoplethysmography bilaterally. Serologic evaluation was unremarkable for autoimmune, malignant, hematologic, and infectious etiologies. Age-appropriate cancer screenings including colonoscopy and low-dose CT chest were negative for malignancy. Patient was reassured and counseled on avoidance of prolonged exposures to the cold.
Conclusions: While generally history and physical examination are focused on differentiating between pernio and acrocyanosis, it is important to remember that the diagnosis of acrocyanosis can be seen concomitantly with pernio.
Poster No. 63
Abstract ID No. 1511241
Venous duplex ultrasound in the diagnosis of venous thoracic outlet syndrome
Merry Ellen D. Barnett, MD, Aditya M. Sharma, MBBS, Randy K. Ramcharitar, MD
University of Virginia
Background: Venous thoracic outlet syndrome (TOS) is the second most common type of TOS, accounting for 3-5% of cases. It is frequently seen in younger men who engage in repetitive upper extremity activity when anatomical variations result in compression of the subclavian vein within the costoclavicular space. We describe a case of venous TOS evaluated at our institution to review the clinical presentation and highlight the utility of ultrasound during diagnosis.
Case presentation: A 50-year-old man presented to vascular medicine clinic for a second opinion on a right upper extremity deep vein thrombosis. He had initially presented to his primary care physician (PCP) seven months prior with pain and edema. At that time, an upper extremity ultrasound showed extensive DVT of the right subclavian, axillary, and brachial veins. The serologic evaluation performed by PCP did not identify an underlying hypercoagulable state and a follow up ultrasound was read as “normal”. He was referred for second opinion on indefinite anticoagulation because he taught physical education, dabbled in body building, and desired to discontinue anticoagulation. A repeat upper extremity venous duplex was notable for normal pulsatile flow in the proximal right subclavian vein and continuous flow at the distal subclavian vein suggestive of more proximal compression or occlusion. CT venogram of the right upper extremity confirmed a 4.5 cm occlusion of the central right subclavian vein as it crossed the medial right clavicle and right first rib on abduction view.
Conclusions: A venous duplex ultrasound performed by an experienced technician can be very helpful for detecting venous stenosis or occlusion and thereby aid in diagnosis of venous thoracic outlet syndrome.
Poster No. 64
Abstract ID No. 1498802
The mysterious case of Mr M
Nichole E. Brunton, DO, Ana Casanegra, MD
Mayo Clinic
Background: Atherosclerosis affects nearly half of the US population. While a common disease, rare complications of advanced atherosclerosis can confound diagnosis. Here, we describe a unique case of moyamoya due to severe intracranial atherosclerosis leading to initial misdiagnosis with antiphospholipid syndrome (APS). His course was complicated by renal failure due to atheroembolism, likely warfarin associated.
Case presentation: A 66-year-old man presented with acute stroke symptoms. Cerebral angiogram demonstrated a moyamoya pattern. Due to persistent symptoms, he underwent direct and indirect revascularization with superficial temporal artery to MCA bypass and encephaloduroarteriosynangiosis. Evaluation revealed a high titer anticardiolipin IgM. Extensive serologic evaluation and PET scan were negative for vasculitis. Due to concern for APS causing moyamoya, anticoagulation was initiated with warfarin. Progressive renal failure developed after the INR was therapeutic. Renal biopsy revealed cholesterol clefts. Given diffuse atherosclerosis, this likely represents warfarin associated atheroembolism. Repeat serology showed rapidly declining anticardiolipin IgM, making APS unlikely. Warfarin was discontinued and he was transitioned to apixaban for new onset atrial fibrillation.
Conclusions: Moyamoya is uncommon and poorly understood. It is associated with a variety of etiologies; careful diagnostic evaluation is necessary to avoid overlooking common etiologies such as advanced atherosclerotic disease.

Severe atherosclerotic disease resulting in Moyamoya and cholesterol emboli, likely warfarin associated.
Poster No. 65
Abstract ID No. 1499073
Aberrant splenic artery aneurysms
Nichole E. Brunton, DO, Fahad Shuja, MBBS, Manju Kalra, MBBS, Ana Casanegra, MD
Mayo Clinic
Background: Splenic artery aneurysms (SAA) are uncommon. Aberrant splenic artery anatomy originating at the superior mesenteric artery is exceedingly rare. Here, we describe a patient with large, complex splenic artery aneurysms occurring concomitantly with aberrant splenic artery anatomy.
Case presentation: A 41-year-old woman with past medical history of 10 vaginal births, one complicated by placental abruption, presents for management of 3 splenic artery aneurysms (measuring 4 cm proximally and 2 cm and 1 cm at the hilum) and mild aortic root dilation (40 mm) incidentally detected on CT imaging after hysterectomy and pelvic reconstruction. Pertinent review of systems includes joint hypermobility (Beighton score 4) alone. No family history of aneurysmal disease or connective tissue disorder. Vascular anatomy survey was completed to exclude involvement of other vascular beds. Genetic counseling and testing were obtained due to suspicion for identifiable heritable connective tissue disorder and remain pending. Due to aberrant anatomy and need for urgent repair of large splenic artery aneurysms, the patient is scheduled for a supraceliac aorta to superior mesenteric artery bypass with splenic artery ligation.
Conclusions: Current guidelines recommend SAA repair when a nonruptured aneurysm reaches ⩾3 cm in size, or at any size in women of childbearing age. Repair is recommended by endovascular approach when feasible, however aberrant splenic artery anatomy with SMA origin poses a unique surgical challenge.

Aberrant splenic artery aneurysms.
Poster No. 66
Abstract ID No. 1499427
The tale of an aortic thrombus
Nichole E. Brunton, DO, Ana Casanegra, MD
Mayo Clinic
Background: Aortic thrombus is rare in patients without aneurysm or atherosclerosis, usually occurring in young patients with prothrombotic disorders. Here, we describe a case of aortic thrombus of unknown etiology resulting in acute limb ischemia and stroke despite anticoagulation.
Case presentation: A 45-year-old woman with history of nicotine use and dyslipidemia presented for spontaneous blue toes. ABI was mildly reduced bilaterally. Vascular imaging revealed an extensive aortic mass versus thrombus involving the aortic arch to descending thoracic aorta. MRA confirmed presence of bland thrombus. Laboratory evaluation was unremarkable for autoimmune, inflammatory, or infectious etiology. Anticoagulation was initiated with heparin and bridged to warfarin therapy on dismissal. She presented one week later for acute onset of right-hand pain secondary to axillary artery occlusion. While en-route to thrombectomy, she developed dysarthria and left hemiparesis. Imaging revealed occlusion of the right ICA with extension to the M1 and M2 segments. Emergent thrombectomy of the ICA with complete recanalization (TICI3) was achieved with neurologic improvement. She subsequently underwent right upper extremity embolectomy with patch angioplasty and clinical improvement in upper extremity symptoms.
Conclusions: Management of aortic thrombus is challenging and optimal treatment is unknown. Large thrombus burden may result in arterial events despite anticoagulation and requires rapid intervention to avoid devastating consequences.

Aortic thrombus, axillary thrombus and internal carotid thrombus.
Poster No. 67
Abstract ID No. 1511599
Köhlmeier-degos disease: an observational analysis of a 16 patient cohort
Cornelia Cudrici, MD1, Natalia Dmitrieva1, Rebecca Huffstutler, CRNP2, Katherine Carney, RN, BSC, CCRC2, Elisa A. Ferrante, PhD1, Marta Cardenas2, Yogen Kanthi, MD3, Douglas Rosing, MD2, Manfred Boehm1
1NIH/NHLBI, 2NHLBI, 3NIH
Background: Köhlmeier-Degos disease (K-D) is a rare small vessel vasculopathy of unknown etiology leading to small blood vessel occlusions in multiple organs, including the skin, central nervous system (CNS), eye, gastrointestinal (GI) tract, lungs, and heart. Approximately 300 K-D cases have been reported in the literature to date. K-D commonly presents as a benign cutaneous form with lesions that appear as erythematous papules and evolve to form a scar with an atrophic, porcelain-white center surrounded by a telangiectatic border. Progression to systemic K-D, or malignant atrophic papulosis, occurs in 2/3 of the patients, is often debilitating and can be fatal (50-70% mortality rate within 2-5 years after diagnosis) due to GI perforations, brain infarcts, spinal lesions, cardiac or pulmonary failure, sepsis or cachexia.
Case presentation: Here, we describe the findings from our single-center longitudinal cohort of 16 patients, nine with systemic KD disease and seven with cutaneous KD. Among patients with systemic involvement, the GI tract is affected in more than followed by CNS involvement. Two of our K-D patients will have both GI and CNS manifestations. In part due to the lack of insight into K-D pathophysiology, no standard of care exists and currently, no clinical trials are available for K-D patients. Eculizumab and treprostinil have been used as off-label treatments for systemic K-D with GI involvement and have shown some moderate-term symptom control. We will present a retrospective analysis of five patients with KD disease treated with treprostinil and eculizumab.
Conclusions: The long-term follow-up on these patients followed at our clinical center over ten years will describe the clinical characteristics of KD patients and evidence of immune dysregulation was commonly observed.
Poster No. 68
Abstract ID No. 1498333
A case of dialysis vascular access (arteriovenous graft) thrombectomy using a novel device Inari InThrill thrombectomy system on an outpatient basis
Rakesh Reddy Devireddy, MD, Osama Qaqi, MD, FACC, FSCAI
Garden City Hospital
Background: Arteriovenous grafts (AVGs) are one of the common vascular accesses used for hemodialysis in patients with End-stage-renal-disease(ESRD). This case describes the successful outpatient-based use of a novel minimally invasive, mechanical thrombectomy device, the InThrill Thrombectomy System (Inari Medical) for the percutaneous thrombectomy of a totally occluded and heavily thrombosed AVG in an ESRD patient on hemodialysis treatment.
Case presentation: A 47-year-old male patient was sent to the outpatient vascular Institute for dialysis access evaluation by angiography with possible intervention. His medical history includes ESRD, on hemodialysis; unable to dialyze due to thrombosed right upper extremity AV graft with hero graft. Prior to the current intervention, the patient underwent interventions for the thrombosis of the same graft on Jan 25, 2023, using Asprex catheter and again on Feb 6, 2023, using Fogarty catheter but presented with recurrent thrombosis of the AV graft. On February 8, 2023, he underwent a successful endovascular revascularization of a totally occluded heavily thrombosed right AV graft with hero graft reduced to less than 20 % residual stenosis/thrombus using Inari Inthrill catheter thrombectomy device and balloon angioplasty resulting in brisk AV graft flow. Patient tolerated the procedure well without complication, was recovered in the holding area with no acute distress/complications. He was discharged the same day after appropriate monitoring on anticoagulant therapy (Rivaroxaban) and was advised to follow up with dialysis appointments per his nephrologist recommendations.
Conclusions: This case report highlights the successful outpatient-based use of the InThrill thrombectomy system for the treatment of thrombosed AV graft in a hemodialysis dependent ESRD patient. To the best of our knowledge there are no reported cases where this device was used on an outpatient basis for percutaneous thrombectomy of the hemodialysis vascular access AV graft. The device was easy to use and efficient. Device and procedure times are unparalleled when compared with thrombolytic-based procedures.

Thrombosed AV graft to Hero graft with InThrill catheter thrombectomy and successful restoration of flow in the AVG with thrombus yield.
Poster No. 69
Abstract ID No. 1493034
A novel treatment for COVID toes/fingers
Andrew B. Dicks, MD, Bruce H. Gray, DO
Prisma Health - University of South Carolina School of Medicine
Background: There has been a myriad of manifestations associated with COVID-19 infections, including skin lesions that resemble pernio which have become known as “COVID toes/fingers.” However, there are limited reports on optimal management of COVID toes/fingers.
Case presentation: A 19-year-old female with no past medical history presents for evaluation of bilateral finger discoloration, ulceration, and pain. The patient reports being diagnosed with COVID-19 one month prior to the onset of her finger symptoms. She initially noted bluish-purple discoloration on the distal aspects of her second and third digits on the right hand. This progressed to involve the first through fourth digits of bilateral hands with associated ulcerations and significant discomfort. She is a non-smoker and denies use of cocaine or ADD medications. She presented to the ED for evaluation three weeks after symptom onset and was given a diagnosis of Raynaud’s disease. Laboratory evaluation was notable for negative ANA, anti-dsDNA, anti-MPO, anti-PR3, centromere B, and SCL-70 antibodies and normal complement levels, ESR, and CRP. She was started on amlodipine 5mg daily and referred to vascular medicine. In the vascular medicine clinic, she reported no improvement in symptoms with amlodipine after one week of therapy. She had persistent discoloration and ulcerations on the distal aspect of her first through fourth digits of bilateral hands (Image A, B). She had normal pulse examination of the upper and lower extremities and no evidence of ulcerations/discolorations of her toes. Upper extremity arterial duplex ultrasound demonstrated dampened but symmetric finger PPG waveforms. Her symptoms were felt to be most consistent with COVID fingers. She was trialed on cilostazol 50mg twice a day and her amlodipine was discontinued. She was seen in follow up one week later with improvement in finger pain and was subsequently followed up one month later with complete resolution of her finger symptoms and healed finger ulcerations.
Conclusions: COVID toes/fingers is a well-recognized complication of COVID-19 infections. However, optimal treatment strategies remain limited. This case report highlights the potential role of cilostazol in patients with COVID toes/fingers.

Poster No. 70
Abstract ID No. 1495317
A rare case of claudication
Jose E. Exaire, MD1, Mohamed Omer, MD2 1Baylor Scott and White, Temple, 2Mayo Clinic
Background: A 40-year old female with past medical history of left iliac DVT secondary to May-Thurner syndrome, presented to vascular clinic complaining of exertional leg pain. She is a nonsmoker and she can barely complete her daily activities due to pain that starts in the right buttock.
Case presentation: A 40-year old female with past medical history of left iliac DVT secondary to May-Thurner syndrome, presented to vascular clinic complaining of exertional leg pain. She is a nonsmoker and she can barely complete her daily activities due to pain that starts in the right buttock and extends to her right calf that improves with rest. Her rest ABI were normal, however, her exercise ABI were markedly abnormal. She was referred to a peripheral arterial disease rehabilitation program where she noticed some improvement from 2.4 METS to 3.4 METS with onset of pain within 5 minutes and resolution after 3 minutes. Given her symptoms, she had a CTA that showed a stent in the left iliac vein origin that impinged on the right iliac artery. An angiogram confirmed the impingement and a rest gradient of 20 mmHg was found, an IVUS showed external compression. She received a covered balloon expanding stent with resolution of the angiographic image as well as normalization of the trans-lesion gradient. Her claudication resolved.
Conclusions: External iliac compression after venous stenting is a rare cause of claudication that should be considered in patients with previous invasive May-Thurner treatment. A high level of suspicious is required. The resting ABI can be normal and further imaging is typically needed.

CTA showing the venous stent impinging the origin of the contralateral artery.
Poster No. 71
Abstract ID No. 1503316
Pulmonary thromboembolism in the intermediate post-operative period of laparoscopic adrenalectomy for an adrenocortical adenoma with Cushing’s syndrome
Ryan Paul A. Fernandez
Makati Medical Center
Background: Cushing’s Syndrome is a constellation of clinical features resulting from chronic exposure to excess glucocorticoids. This is suggested to induce hypercoagulability, which results in a 10-fold increase in the risk for venous thromboembolic events. It results from an increased production of procoagulant factors and impaired fibrinolytic, leading to decreased activated partial thromboplastin time and increased clot lysis time. However, reported events are quite rare and majority of which occur in the post-operative setting. A systematic review done in 2009 by Zaane, et. al., discussed that there is up to a 1.5% incidence of venous thromboembolism after transsphenoidal surgery and 0.3-3% after laparoscopic adrenalectomy. Despite this, it is still up to debate whether thromboprophylaxis should be instituted post-operatively among these patients.
Case presentation: The case is a 42-year-old Filipino, female, with Cushing’s Syndrome readmitted because of generalized body weakness, seven days after laparoscopic adrenalectomy of a left adrenocortical adenoma. After correction of electrolytes and further evaluation, findings suggested the possibility of an infection of the post-operative site. This was confirmed with a whole abdominal CT scan with contrast. She was then managed conservatively with antibiotics. The study was repeated four days after for surveillance of the abscess site. It showed incidental findings of diffuse thromboembolism on the right main pulmonary artery and its segmental branches, right common iliac vein, internal iliac vein, superior gluteal vein, and left ovarian vein. No hypotension, dyspnea, syncope, leg pain, or swelling was noted during this period. Findings were confirmed through CT Angiography of Pulmonary Arteries. Furthermore, no right ventricular dysfunction was noted on 2D echo. She was then treated with loading dose of Rivaroxaban and eventually sent home well after resolution of infection and maintained on anticoagulation.
Conclusions: Cushing’s syndrome compounded by post-operative immobility is a significant risk factor in the development of pulmonary thromboembolism despite the absence of other risk factors contributing to its development.
Poster No. 72
Abstract ID No. 1498345
An unusual cutaneous reaction to anticoagulation: a case of recurrent hemorrhagic bullae from enoxaparin and then fondaparinux
Atefeh Ghorbanzadeh, MD, Damon Houghton, MD, MS, Emily Bendel, MD
Mayo Clinic
Background: Bullous hemorrhagic dermatosis is an uncommon, non-immune, self-limiting drug reaction that can occur mostly after the use of unfractionated heparin (UFH) and low-molecular-weight heparin (LMWH), and in rarer instances, caused by other anticoagulants.
Case presentation: We present a case of an 83-year-old male with multiple multimorbidities, including mechanical replacement of the aortic and mitral valves with Saint Jude valves in 2003, persistent atrial fibrillation diagnosed in 1993 with a total of six cardioversions, mixed ischemic/nonischemic cardiomyopathy with an ejection fraction of 28% post-defibrillator placement/PPM biventricular, transient ischemic attack, COPD, Paget’s disease involving the scrotum area, and prostate cancer (in remission). He has taken warfarin (target INR 2.5-3.5) since 2003 due to a history of cardiac valve surgery. In July 2021, he started taking enoxaparin in order to bridge to warfarin for a subtherapeutic INR, and after receiving five injections of 100 mg of enoxaparin, he developed lesions all over his body (fig 1). Accordingly, he was advised to avoid using all forms of heparin due to concern for heparin-induced skin necrosis (there was clinically apparent thrombosis or thrombocytopenia). In February 2023, the patient had another subtherapeutic INR of 1.90 and was prescribed fondaparinux sodium 7.5 mg subcutaneously for three days. He immediately developed again tense hemorrhagic bullae on the left arm, elbow, and shin neither painful nor pruritic, similar to those observed after enoxaparin administration (fig 2). Apart from these findings, no petechiae or other signs of bleeding were observed. Both incidences were resolved once the medication was stopped. Except for the long aPTT of 47 seconds (reference 25 - 37 sec), the INR of 3.8 (reference 0.9 - 1.1), and the PT of 44.6 (reference 9.4 - 12.5 sec), the other coagulation profiles, including platelet count, heparin platelet factor 4, and serotonin release assay, were within normal ranges.
Conclusions: In conclusion, Hemorrhagic bullae are rare adverse reactions to heparin-based anticoagulants, which are benign conditions that do not affect coagulation tests or platelet counts, and only require monitoring until the rash has resolved.

Fig 1. Hemorrhagic blisters on upper extremity overlying normal skin after taking LMWH (enoxaparin), Fig 2. Hemorrhagic blisters on lower extremities overlying normal skin after fondaparinux.
Poster No. 73
Abstract ID No. 1494460
Atypical presentation of Lemierre’s syndrome - septic emboli to brain and liver caused by Fusobacterium varium
Melissa Hidalgo, MD, Sophia Navajas, MD, Laura Miranda, MD, Noha Abdelgelil, MD, Enoch Johnmarie, MD, Fares Qureiyeh, MD
Broward Health North
Background: Initially described in 1936, Lemierre’s syndrome (LS), also known as post-anginal septicemia, classically presents in young healthy adults age 14 to 24. LS is characterized by bacteremia and thrombophlebitis caused by Fusobacterium (F) necrophorum, F. nucleatum, and most recently (2016) by F. varium species, which are gram-negative anaerobic bacteria found in oropharyngeal and gastrointestinal flora. We report an atypical presentation of LS.
Case presentation: 63-year-old male with medical history of hyperlipidemia brought to the emergency room with altered mental status; last seen normal 3 days prior. On admission, vitals showed 100.4 F, 155/91 mmHg, 100 bpm, 24 br/min. On physical exam, regular rate and rhythm, reactive pupils, corneal and gag reflex intact, obtunded, all extremities moved spontaneously, did not follow commands; intubated for airway protection. EKG was unremarkable. Labs were significant for WBC 31.73, AST 143 unit/L, ALT 226 unit/L, alkaline phosphatase 161 unit/L, total bilirubin 2.3 mg/dL, LDH 424 unit/L, D-dimer 3.70 ug/mL FEU, creatine kinase 1089 unit/L, urine toxicology unremarkable. On imaging, CT brain had no acute findings, CT abdomen had complex cystic liver lesions up to 7.7 cm, liver ultrasound showed deep venous thrombosis in inferior vena cava. MRI brain revealed supra- and infratentorial microabscesses with punctate hemorrhage. Broad spectrum antibiotics were started. Hepatic abscesses were drained. CSF, sputum, urine, hepatic abscess fluid, hepatitis, tuberculosis, HIV, Entamoeba histolytica studies were negative. Blood cultures grew F. varium on day 8. Subsequently, CT neck revealed 4.3 cm right mandibular abscess with no thrombosis. Unfortunately, prognosis was poor and patient was discharged to hospice.
Conclusions: Atypical LS with disseminated septic emboli remains rare. Diagnosis is confirmed via imaging as blood cultures take 5-8 days. Treatment includes 4-6 weeks of antibiotics, occasionally combined with anticoagulation and surgical intervention. LS is associated with a delay in diagnosis and mortality up to 18% even with appropriate medical therapy. Thus, early diagnosis and treatment are crucial to reduce morbidity and mortality rates.
Poster No. 74
Abstract ID No. 1498303
Infected common femoral artery pseudoaneurysm due to heterotopic ossification
William Kelly, BS1, Joel Keefe, BS1, Brian Jones, MD2, David Chew, MD2, Chris Carsten, MD2
1University of South Carolina School of Medicine, 2Prisma Health-University of South Carolina
Background: Hip fractures are an increasingly common injury in the aging population and are often attributed to complications following injury and surgical fixation. Heterotopic ossification (HO) is the formation of mature bone where bone does not normally exist, and can be seen in patients who undergo open reduction and internal fixation (ORIF) of a hip fracture. Rarely, HO can cause injury to nearby vascular structures and may require surgical intervention.
Case presentation: A 63-year-old male with a past medical history significant for alcohol use disorder and right hip fracture with surgical fixation two years ago presented to an outside hospital for right upper leg pain. A CT scan revealed HO of the proximal right femur and pseudoaneurysm of the right common femoral artery. Distal pulses were intact, and a pulsatile mass was present in the right groin. The patient was transferred to a tertiary care center for evaluation by vascular surgery and medically optimized. He then underwent surgery to excise the aneurysm and the heterotopic bone which appeared to cause the injury. A cadaveric arterial graft and sartorius flap were used for the closure. Wound cultures revealed bacterial growth from both the artery and the excised bone.
Conclusions: The presentation of pseudoaneurysm after a nearby fracture varies widely in the literature. Vascular injury often presents immediately after ORIF due to bone fragments or sharp surgical instruments contacting a vessel during manipulation of the fracture. Due to its course close to the femur, the deep femoral artery is more often injured than the common femoral artery, making this a more rare presentation. Pseudoaneurysms form due to vascular wall injury, causing an outpouching that is enclosed by products of the clotting cascade, instead of being enclosed by the vessel wall like a true aneurysm. Often they are caused by endovascular procedures, but may be due to trauma similar to this case. The injured vessel may become a source of bacterial infection, and these complicated cases should be treated with excision and repair.
Poster No. 75
Abstract ID No. 1511279
A rare cause of cervical radiculopathy: dissecting pseudoaneurysm
Bilal Bucak, MD, Carmen R. Holmes, MD, Ana Casanegra, MD, Giuseppe Lanzino, MD, Zafer Keser, MD
Mayo Clinic
Background: Cervical vertebral artery dissection is a rare condition (2 in 100,000/year). Twenty percent of patients develop pseudoaneurysm, which can exert mass effect over surrounding tissues. The incidence of radiculopathy in patients with cervical vertebral artery dissection or dissecting pseudoaneurysm is estimated to be less than 1%. Given potentially devastating complications and different, treatment than other causes of cervical radiculopathy, it is crucial to include dissecting pseudoaneurysm in the differential diagnosis.
Case presentation: 34 years-old male patient with a personal history of Loeys-Dietz syndrome presented with sudden onset radicular left shoulder pain and abduction weakness compatible with C5 radiculopathy. Initial cervical MRI without contrast showed a mass of a likely vascular origin compressing over the left C5 root. A subsequent CT Angiogram showed a left vertebral artery dissecting a pseudoaneurysm of 11 mm in size protruding into the cervical canal. We decided to conservatively manage the dissecting pseudoaneurysm with medical treatment (low-dose daily aspirin). Pseudoaneurysm showed spontaneous regression with complete resolution of symptoms in 3 months.
Conclusions: We present a unique case of cervical radiculopathy caused by vertebral artery dissecting pseudoaneurysm. In patients with underlying connective tissue disease, such as Loeys-Dietz syndrome, vascular Ehlers-Danlos syndrome, and Marfan syndrome, dissecting pseudoaneurysm should be considered in the differential diagnosis. Given potentially devastating complications and different management strategies, it is essential to include vertebral artery dissecting pseudoaneurysm in the differential diagnosis of cervical radiculopathy, especially when routine cervical imaging raises suspicion of vascular pathology. When to utilize endovascular treatment in dissecting pseudoaneurysm remains controversial. In our case, the patient had spontaneous regression of his pseudoaneurysm without intervention. Our case suggests conservative management initially with endovascular intervention as second-line treatment as needed basis might be a reasonable strategy.

Poster No. 76
Abstract ID No. 1497212
Coronary aneurysm with intraluminal thrombosis after spontaneous coronary artery dissection
Muhammad Umar Khalid, MD1, Meghann McCarthy, DO1, Emmanuel Akintoye, MD1, Pulkit Chaudhury, MD1
1 Cleveland Clinic Foundation
Background: Spontaneous coronary artery dissection (SCAD) is a rare, yet increasingly recognized cause of myocardial infarction in young patients with minimal atherosclerotic vascular disease. Complications of SCAD include ischemic cardiomyopathy, cardiogenic shock, ventricular arrhythmias and rarely cardiovascular death. We present a unique case of SCAD complicated with coronary artery aneurysm.
Case presentation: A 23-year old G5P4 female presented 3 weeks postpartum with acute onset chest pain. Electrocardiogram revealed ST depression and T-wave inversions in the precordial leads. Troponin I was 1.46 ng/mL and echocardiogram was significant for global LV hypokinesis. Coronary angiogram revealed a type 1 dissection of the left main coronary artery (LMCA) extending into the LAD and LCx. A type 2 RCA dissection was also seen. Coronary artery bypass grafting was deferred by the patient. Further imaging and genetic testing were negative for other arteriopathies. Patient presented 3 years later with stable angina and CT revealed a LMCA aneurysm with intraluminal thrombus resulting in severe stenosis. Patient again deferred surgery and was started on warfarin. At the 4-month visit, she reported resolution of symptoms and CT revealed decrease in thrombus burden.
Conclusions: Coronary aneurysms after SCAD are rare and not well studied. Coronary aneurysms in other conditions such as Kawasaki disease are associated with thrombosis, rupture and death. Oral anticoagulation can be considered for management of intraluminal thrombosis.

Coronary aneurysm and intraluminal thrombosis after SCAD.
Poster No. 77
Abstract ID No. 1511502
Granulomatosis with polyangiitis masquerading as thromboangiitis obliterans
Muhammad Umar Khalid, MD, Teresa Wu, MD
Cleveland Clinic Foundation
Background: Granulomatosis with polyangiitis (GPA) is an antineutrophil cytoplasmic autoantibody (ANCA) associated necrotizing vasculitis affecting small-sized arteries. While cutaneous manifestations are present in approximately 20% of cases, digital ischemia is rare, and may be attributed to other causes. We present a unique case of GPA with digital ischemia that was initially diagnosed as Thromboangiitis Obliterans (TAO).
Case presentation: A 48 year old male with a history of chronic polyarthralgias, hyperlipidemia, hypertension and a 60 pack year smoking history presented with black discoloration of the left third and right first digits and left hallux. Laboratory testing revealed mildly elevated ESR and CRP. c-ANCA/PR3 were positive, however suspicion for GPA was low given lack of other signs and symptoms of vasculitis. Upper extremity angiogram revealed multiple proximal and mid occlusions of the digital arteries. Given his extensive history of smoking, TAO was suspected and the patient was started on amlodipine and counseled on smoking cessation. One mother later, he presented with fevers, weight loss, sinus fullness, left foot drop and right leg weakness. Laboratory testing revealed markedly elevated ESR and CRP, and EMG demonstrated left sciatic axonal mononeuropathy. Given these findings, the patient was ultimately diagnosed with GPA and started on methylprednisolone with improvement in symptoms.
Conclusions: Digital ischemia is a rare complication of GPA, and may quickly be attributed to other causes. Extensive tobacco exposure made the diagnosis of GPA vs TAO difficult in this case. A high index of suspicion is critical for prompt recognition and treatment in order to minimize complications.
Poster No. 78
Abstract ID No. 1506367
Endovascular interventions for IVC agenesis and atresia
Dhara Kinariwala, MD1, Minhaj S. Khaja, MD, MBA, FSIR, FSVM2, David Williams, MD, FSIR2
1University of Virginia, 2University of Michigan2
Background: Inferior vena cava (IVC) atresia/agenesis comprises a highly variable set of conditions in which the venous system either fails to develop normally or degenerates prematurely. These conditions can be asymptomatic or have varying manifestations, including deep venous thrombus, post-thrombotic syndrome, pelvic venous disease at all stages of life. Treatment depends on the patient’s symptoms, and may include anticoagulation and medical management, surgical reconstruction, and endovascular recanalization.
Case presentation: A literature review was performed on endovascular treatments of various IVC abnormalities with emphasis on atresia & agenesis. In addition, successful endovascular treatments of IVC occlusions were compiled per multicenter experience, including workup and post-intervention care. Visual, angiographic data was organized using embryologic and anatomic illustrations for ease of explanation. Endovascular options for treatment of IVC agenesis vary and may include thrombolysis or recanalization/reconstruction. Thrombolysis has been described for treatment of IVC agenesis-associated DVT, both in isolation and in coordination with recanalization as a staged endovascular procedure. For recanalization, blunt or sharp recanalization is used with imaging in orthogonal planes, followed by balloon angioplasty and stenting. Adequate inflow is crucial to maintain patency. Long-term anticoagulation is generally recommended for IVCA, although there is no consensus on type and dosage. Sequential compression devices and activity, generally walking multiple times per day, is recommended.
Conclusions: IVC atresia/agenesis is an embryologically complex process thought to occur due to insult to the formed or forming IVC. This entity is often asymptomatic until adulthood, and may present with DVT and chronic venous insufficiency. Management includes intervention and/or surgery, anticoagulation, and compression, although no specific consensus exists. Further studies are necessary to establish rates of technical success of endovascular management, long-term patency, and clinical outcomes, as well as provide standardized recommendations on treatment approaches and goals for these patients.
Poster No. 79
Abstract ID No. 1499907
A rare genetic cause of cerebral aneurysms and strokes in a young woman with a family history of psychological disturbances
Isabelle Kornblau, MS, CGC, Veronica Fettig, MS, CGC, Christopher P. Kellner, MD, Hazem Shoirah, MD, Gesille Fiallo, P, Amy Kontorovich, MD, PhD6
Icahn School of Medicine at Mount Sinai
Background: Moyamoya disease (MMD) is a rare vaso-occlusive disease characterized by progressive stenosis of the intracranial arteries and compensatory creation of small vessel networks (known as moyamoya) for collateral circulation. Cerebrovascular complications include ischemic stroke, cerebral aneurysms, moyamoya collateral ruptures, and intracranial hemorrhages that may arise in childhood or adolescence.
Case presentation: We describe a 30-year-old woman with a history of multiple arterial stenoses and aneurysms, stroke, and migraines, as well as a paternal family history of psychiatric and behavioral abnormalities including violence, and severe migraines. Recently, the patient started exhibiting symptoms of impulsivity and verbal aggression. Genetic testing revealed a heterozygous RNF213 polymorphism (p.R4810K), indicative of MMD.
Conclusions: The MMD diagnosis provided an explanation for our patient’s multiple intracranial stenoses, strokes, and cerebral aneurysms and dissections, as well as her longstanding history of migraines. Migraines may stem from dilated transdural collaterals or from cerebral hypoperfusion. Aggressiveness could be explained by frontal lobe infarcts from occlusive-mediated ischemic episodes in the context of MMD. Psychiatric comorbidities similar to those seen in the patient’s family history, such as agitation, have previously been noted in numerous MMD case reports. We present this MMD case as illustrative of a rare but possible genetic cause of migraines, strokes, and cerebral aneurysms in young adults with a personal or family history of behavioral/psychological disturbances. In such cases, MMD should be considered as part of the diagnostic differential, especially in East Asian patients. Molecular diagnosis can identify other at-risk family members and aid in the establishment of screening of cerebral aneurysms and stenosis. Surgical revascularization procedures can be used to reduce rates of future strokes. Additional conventional regimens like control of hypertension, statin therapy for hyperlipidemia, and antiplatelet therapy following diagnosis can lower the rate of complications and subsequently lead to better cardiovascular and neurocognitive outcomes.

Moyamoya disease family pedigree.
Poster No. 80
Abstract ID No. 1496054
Addressing the root: a case report on the recognition and management of May-Thurner syndrome in an elderly male
Marie Antoinette A. Lacson, M.D.1, Marilou de Jesus, M.D.1, Fabio Enrique B. Posas2, Precious Samonte2
1St. Luke’s Medical Center-Global City1, 2St. Luke’s Medical Center- Bonifacio Global City
Background: May Thurner syndrome (MTS) is a rare vascular compression syndrome of the iliac veins accounting for 2-5% of deep venous thrombosis (DVT) cases. Commonly seen in young females and rarely reported in elderly males. This paper highlights the significance of recognizing MTS in an elderly male presenting with persistent unilateral DVT despite adequate anticoagulation. Further work ups should be pursued in order to identify this entity and institute definite management.
Case presentation: A 71-year-old male presents with a one year history of progressive swelling of the left lower extremity. He was diagnosed on ultrasound with extensive DVT from the left common iliac vein (CIV) down to the popliteal veins and managed with therapeutic low molecular weight heparin for two weeks but no improvement. CT venography was done which showed an ectatic right common iliac artery compressing the left common iliac vein. The patient underwent inferior vena cava filter placement, venous stenting of the left CIV, and catheter-directed thrombolysis. There was clinically significant improvement with decrease in leg swelling. Follow-up ultrasound done showed reconstitution of flow in the left proximal venous segments.
Conclusions: MTS should be considered in an elderly male with unilateral extensive DVT. Studies support catheter-directed thrombolysis and anticoagulation to be superior to anticoagulation alone in iliofemoral venous thrombosis secondary to MTS and iliac venous stenting to relieve obstruction.

Left: Doppler ultrasound showing left common iliac vein compressed by an ectatic right common iliac artery. Right: Left common iliac vein stent.
Poster No. 81
Abstract ID No. 1473992
Total occlusion of the abdominal aorta: a case of Leriche syndrome
Kirsten M. Lipps, MD, Raymond C. Shields, MD
Mayo Clinic
Background: Leriche syndrome is an uncommon, severe manifestation of peripheral artery disease (PAD), which results in total occlusion of the abdominal aorta (AA). It is characterized by a triad of proximal lower extremity claudication, erectile dysfunction (ED), and diminished or absent femoral pulses.
Case presentation: A 73 year-old man with coronary artery disease, ED, tobacco use, and colon cancer presented with longstanding buttock and thigh pain with exertion. He had severely diminished femoral pulses and absent distal pulses. Bilateral resting ankle-branchial indices (ABIs) were 0.3, consistent with severe PAD. Computed tomographic angiography demonstrated total occlusion of the infra-renal AA and bilateral common iliac arteries (Figure 1). Given symptom severity and disease complexity, surgical aortobifemoral bypass was the preferred revascularization approach. However, this was not pursued given need to expedite healing to minimize chemotherapy interruption. Axillobifemoral bypass was also not chosen given risk of infection with an extra-anatomic bypass in the setting of immunosuppression. Consequently, the patient underwent percutaneous Covered Endovascular Reconstruction of Aortic Bifurcation (CERAB) (Figures 2 and 3). Post-operatively, the patient’s symptoms resolved, and ABIs normalized.
Conclusions: Leriche syndrome may be misdiagnosed due to lack of typical distal leg claudication. CERAB is a minimally invasive revascularization technique that is safe and effective for aortoiliac occlusive disease.

Poster No. 82
Abstract ID No. 1493348
Multimodality imaging to differentiate intramural hematoma mimicking aortitis
Tabitha N. Lobo, MD1, DIllon Gibson, MD1, Pablo Ruda Vega, MD1, Santosh Bhusal, MD1, Joshua Clevenger, MD2, Francis Lytle, MD1, Sanjay Rajagopalan, MD2, Teresa L Carman, MD2, Heather L. Gornik, MD, MSVM2
1University Hospitals Cleveland Medical Center, 2University Hospitals Harrington Heart & Vascular Institute
Background: Similar radiological findings can result in a diagnostic conundrum while differentiating aortitis from intramural hematoma (IMH), two entities with different management strategies.
Case presentation: A 59-year-old female with no significant past medical history presented with epigastric and chest pain, and a blood pressure of 227/116 mmHg. She was an active smoker, and reported fever, chills and malaise. Urgent CT angiogram revealed circumferential soft tissue density surrounding the thoracic aorta from the distal arch to the bifurcation of iliac arteries, concerning for aortitis. There was similar soft tissue density surrounding the branches of the celiac artery with pleural and pericardial effusions. ESR was 81 mm/h and CRP was 16 mg/dL. She was started on esmolol drip for blood pressure control and prednisone at 1mg/kg for aortitis. Eventual infectious and rheumatological work up was negative. PET/CT showed mild FDG avidity associated with circumferential hyperdensity surrounding the aorta concerning for inflammation. MRA sequentially demonstrated diffuse circumferential thickening of the aorta with heterogenous signal characteristics concerning for a fresh thrombus that extended from the origin of the left subclavian artery to slightly below the renal arteries confirming the diagnosis of extensive type B IMH. Steroids were discontinued. She transitioned to an oral hypertensive regimen with good control of her pain and a plan to repeat imaging in 3-6 months.
Conclusions: With the management of these two aortic pathologies being highly reliant on radiographic evidence, multimodality imaging can play a pivotal role in differentiating diffuse IMH from aortitis.

Sagittal (A) and Transverse (B) CT angiogram views depicting circumferential hyperattenuating soft tissue density surrounding the descending aorta till the bifurcation of iliac arteries favoring a diagnosis of aortitis. Sagittal (C) and Transverse (D) PET/CT views showing mild FDG avidity with circumferential hyperdensity surrounding the aorta concerning for inflammation. Sagittal (E) and Transverse (F) MR angiogram views showing diffuse circumferential thickening of the descending aorta from the origin of the left subclavian artery to slightly below the renal arteries with heterogenous signal characteristics concerning for fresh thrombus, suggestive of intramural hematoma.
Poster No. 83
Abstract ID No. 1476513
Aortoiliac occlusive disease (Leriche syndrome) due to embolization of the left ventricular thrombus from methamphetamine-induced cardiomyopathy
Kaye Eunice L. Lustestica, Nigel Jeronimo Santos, Pocholo Carlo Bernardo, Ace Samson, Muriel A. Morilla-Buco
Philippine General Hospital
Background: Leriche syndrome also known as aortoiliac occlusive disease (AIOD) is a devastating presentation peripheral arterial disease (PAD) involving the distal abdominal aorta and iliac arteries. It is composed of a triad of intermittent claudication, erectile dysfunction and decreased or absent distal pulses. Prevalence of aortoiliac occlusive disease in general population ranges from 4.56% to 14% .
Case presentation: A 31-year-old Filipino male with no known comorbidities, but with exertional dyspnea on ordinary activities. He is a 20-pack year smoker with history of methamphetamine use. He presented with a 2 month history of gradual progression of bilateral lower extremity weakness associated initially with claudication and loss of sensation which progressed to erectile dysfunction, pulselessness and limb discoloration involving bilateral thigh down to the feet. CT Aortogram was done showing an aorta with its lumen opacified by contrast down to the level of L2 and at the level of L3 with complete occlusion of the aorta. 2Decho was done showing multiple echogenic densities in the left ventricular apex the largest of which measures 1.0mm x 2.9 mm and the other two measuring 1.0 mm x 1.0 mm and 0.9 mm x 1.1 mm respectively. Patient underwent embolectomy of the infrarenal abdominal aorta and bilateral iliac system and bilateral hip disarticulation. Histopathologic diagnosis of the embolus is fibrinohemorrhagic material consistent with embolus. Unfortunately patient expired due to sepsis from hospital acquired pneumonia.
Conclusions: Aortoiliac occlusive disease can be catastrophic due to high mortality rate. Emergent procedure of endovascular or surgical revascularization with the most instantaneous restoration of arterial blood flow and minimum possible risk should be accomplished. To provide the best quality post-operative care is also one of utmost priority to improve outcome. Interdisciplinary subspecialty care team should include highly-skilled cardiovascular medical and surgical physicians.

CT aortogram showing complete thrombosis of the aorta at the iliac bifurcation.
Poster No. 84
Abstract ID No. 1495063
Notch pathways in the vascuar medicine clinic: more than just the pulmonary artery
Gretchen MacCarrick, MS, CGC1, Elizabeth Ratchford, MD1, Philip Seo, MD1, Harry C. Dietz, MD2
1Johns Hopkins School of Medicine, 2Johns Hopkins University
Background: NOTCH signaling is critically involved in the regulation of vascular smooth muscle cell phenotypic switching (contractile-to-proliferative). In humans, 4 different NOTCH receptors exist, comprising NOTCH1-4 that interact with ligands in the Delta and Jagged (JAG) family. The pathway has been implicated in the genesis of pulmonary arterial hypertension. Pathogenic variants in human NOTCH signaling genes have also been identified in different types of cardiovascular disease (bicuspid aortic valve, left-sided congenital heart disease (CHD), pulmonary valve stenosis) in Alagille and Adams-Oliver syndromes. There are also increasing reports of intracranial, thoracic and visceral artery disease associated with this pathway. Here we present two multi-generational families with possible NOTCH-related vascular disease, as well as review of aneurysm disease and CHD in NOTCH-related conditions.
Case presentation: Family 1 underwent whole exome sequencing and was found to have a variant of uncertain significance (VUS) in JAG1, specifically c.755G>A, in all three affected family members. This variant has been reported in a patient with Alagille syndrome. Family members are variably affected by pulmonary and visceral artery stenoses as well as, thoracic aortic and intracranial aneurysm, and CHDs. Family 2 underwent gene panel testing and was found to have a VUS in the NOTCH3 gene, specifically c.802+6C>G. This variant is detected in population databases in ~1 in 11,000 people of European ancestry, and less likely to be cause of disease in this family, as there is not correct familial segregation. Thus, this family is an excellent candidate for whole exome sequencing. The proband experienced a subarachnoid hemorrhage at age 29 and had 6 intracranial aneurysms clipped and ophthalmic artery wrapping. Subsequently she had mesenteric artery aneurysms, with 5 requiring coiling and stenting. Other family members have shown carotid and visceral artery aneurysms and stenoses.
Conclusions: The variability of CHD and aneurysm presentations in patients with NOTCH-related disease indicate that these genes should be included in any aortopathy or arterial disease gene testing strategy.
Poster No. 85
Abstract ID No. 1511327
Catch me if you can: successful right atrial thrombectomy and pulmonary embolectomy in a young female with intracardiac type a thrombus in transit and a massive pulmonary embolism. A case report
Roi Joseph Mendoza, Flordeliz Lontok, Marilou de Jesus, MD
St. Lukes Medical Center-Bonifactio Global City
Background: Right atrial thrombus in transit leading to pulmonary embolism is a rare disease associated with high mortality. Optimal management remains controversial but a pulmonary embolism response team increases the chances of survival. We aim to present an example of this disease with a favorable outcome.
Case presentation: A 36-year-old Filipino female presented at the ER due to left leg swelling and loss of consciousness. She was hypotensive, tachycardic, tachypneic, had low oxygen saturations, and required inotropic support. 2D echocardiogram showed a type A right atrial thrombus in transit, dilated right atrium, and right ventricle. CTPA confirmed the presence of a massive pulmonary embolism, and a venous duplex scan showed extensive DVT on the left lower extremity. She underwent right atrial thrombectomy, pulmonary embolectomy, and inferior vena cava filter insertion with improvement of hemodynamics. She was discharged stable on anticoagulant and continued to improve on follow-up.
Conclusions: Right atrial surgical thrombectomy with pulmonary embolectomy is a viable option in the management of right atrial thrombus in transit with massive pulmonary embolism. A pulmonary embolism response team further increases the chances of survival of these patients. However, further studies are needed to determine optimal treatment strategies for the management of these cases.

Apical focused right ventricle view on 2D echocardiogram showing an echogenic density (white arrow) traversing the tricuspid valve during diastole. Right ventricle (RV), right atrium (RA), left ventricle (LV), left atrium (LA)
Poster No. 86
Abstract ID No. 1511383
Small bite big threat: digital ischemia in a patient suffering from severe Plasmodium falciparum infection complicated by disseminated intravascular coagulation
Roi Joseph Mendoza, Diana Jean Roxas, Marilou de Jesus, MD
St. Lukes Medical Center-Bonifactio Global City
Background: Digital ischemia is a rare complication of Plasmodium falciparum infection with high rates of amputation. We present a case of a 36-year-old female who had a recent travel in South Sudan and was infected with a severe form of malaria complicated by digital ischemia, disseminated intravascular coagulation (DIC), and impending acute respiratory distress syndrome.
Case presentation: A 36-year-old Filipino female who works as a humanitarian volunteer in South Sudan went back to the Philippines. Three days after, she complained of fever and presented to the ER jaundiced. Workup was consistent with severe malaria with heavy parasitemia on blood smear. She was immediately started on anti-malarial regimen but complications such as DIC and impending ARDS was already present. On the second hospital day, symptoms worsened now with hypoglycemic episodes and examination showed discoloration in all digits of the bilateral lower extremities with pain and numbness. Low dose enoxaparin was started despite presence of DIC, bleeding parameters were monitored closely, supportive therapy given and on the 8th day of anti-malarial regimen patient slowly improved including the digital ischemia and eventually discharged
Conclusions: Malaria is still a common disease in some parts of the world including South East Asia. A rare complication includes digital ischemia which has a high rate of amputation hence we must consider the risk and benefits of anticoagulation especially in patient with DIC. Early recognition and prompt treatment may help prevent limb loss.

Digital ischemia in all digits of the bilateral lower extremities in a patient suffering from severe malaria complicated by disseminated intravascular coagulation.
Poster No. 87
Abstract ID No. 1476339
Unraveling what lies beneath: a rare case of May-Thurner syndrome variant and arteriovenous fistula in an octogenarian female
Avianne Krystle S. Mendoza, MD, Marilou de Jesus, MD
St. Luke’s Medical Center Global City
Background: May-Thurner syndrome (MTS) manifests as unilateral lower extremity edema, pain, varicosities, and venous ulcers due to iliac vein compression. This may cause deep vein thrombosis (DVT) and chronic venous insufficiency. MTS related DVTs occur in 2% to 3% of all cases, usually seen in young females and rarely in elderly. Most common areas of compression involves the common iliac vein and ipsilateral arteries but other variants were reported.
Case presentation: This is a case of an 89-year-old female with persistent left lower extremity swelling, recurrent DVT and post-thrombotic syndrome (PTS). She underwent left internal iliac artery coil embolization due to presence of an arteriovenous fistula. Further work ups showed compression of the left common iliac vein by the left common iliac artery illustrating a May-Thurner Syndrome variant. This may have led to peripheral venous hypertension and arteriovenous fistula formation. Palma procedure (femoral crossover bypass surgery) was done and significant improvement in patient’s overall condition was achieved.
Conclusions: MTS can lead to recurrent DVT and PTS if left unrecognized and untreated. MTS must be considered in patients with chronic unilateral limb swelling. Endovascular approach is the gold standard of treatment but, venous bypass is an equally effective treatment option that can improve limb function and quality of life.

Poster No. 88
Abstract ID No. 1475472
A hostile environment: thoracoabdominal aneurysm degeneration in a patient with uncontrolled HIV/AIDS
Amanda M. Morrison, MD, Alexander E. Sullivan, MD, Aaron W. Aday, MD, MSc
Vanderbilt University Medical Center
Background: A 39-year-old man with a remote history of IV drug use presented with abdominal pain. Imaging showed a thoracoabdominal aneurysm measuring up to 5.5 cm (Figure) with an inflammatory-appearing rind. His course was complicated by MSSA bacteremia and new diagnoses of HIV (CD4 34 cells/µL) and hepatitis B. His aneurysm was felt to be of infectious etiology, though lymphoma and peri-aortic fibrosis were also considered. He was not felt to be an operative candidate due to his comorbidities and was scheduled for outpatient follow up. Genetic testing revealed no pathogenic variants.
Case presentation: He was subsequently lost to follow up for 5 years before representing with dyspnea and orthopnea. Lab work demonstrated lack of viral suppression and low CD4 count (< 50 cells/µL). Imaging revealed progressive dilatation of his aneurysm, now measuring 7.7 x 7.5 cm in the abdominal aorta (Figure). Transthoracic echocardiography demonstrated ejection fraction (EF) 45-50%, severe aortic regurgitation, and right ventricular dysfunction. A staged surgical repair was planned given his valvular disease and low-normal EF. He underwent valve-sparing aortic root replacement and ascending aortic aneurysm repair; pathology demonstrated marked lymphocytic infiltration in the outer vessel wall and adventitia with severe atherosclerosis and fibrosis, consistent with chronic inflammation. Following recovery, he underwent combined open thoracic and abdominal aortic aneurysm repair. His operation was complicated by RV dysfunction, liver failure, and coagulopathy, and he died shortly thereafter.
Conclusions: This is a case of progressive thoracoabdominal aortic aneurysm degeneration in the setting of uncontrolled HIV/AIDS. Though the relationship between HIV and aneurysmal disease is incompletely understood, chronic inflammation related to unsuppressed HIV is a risk factor for aneurysm progression.

Figure. A, CT angiogram on initial presentation showing the thoracoabdominal aneurysm with abdominal aortic measurements of 5.5 x 5.2 cm (arrows) and an inflammatory rind (asterisk). B, Subsequent chest CT angiogram demonstrating an ascending aortic aneurysm measuring 5.3 cm (closed arrow) and a descending thoracic aortic aneurysm measuring 6.3 x 5.6 cm (arrows). C, CT angiogram also demonstrated an abdominal aortic aneurysm of 7.7 x 7.5 cm (arrows).
Poster No. 89
Abstract ID No. 1499048
Carotid artery dissection - an ominous complication in patients with acute type a aortic dissection: a case series
Nyssa Elline Palileo, MD, FPCP, FPCC, Donna Aurea Maderazo, MD, Matt Viriña, MD, Lea Arceli Gonzales Porciuncula
St. Luke’s Medical Center - Quezon City
Background: Carotid artery involvement demonstrated an incidence of 30% in acute type A aortic dissection and is a predisposing factor to irreversible cerebral malperfusion. Although data suggest concomitant endovascular repair of common carotid artery dissection during open ascending aortic repair, the criteria for repair, natural history and risk of stroke are unclear.
Case presentation: This report discusses three cases of acute type A aortic dissection with carotid artery extension. Case 1 was a 48 year old female with sudden onset of severe “tearing” chest pain radiating to the back. CT aortogram revealed Stanford A, Debakey I dissection from the sino-tubular junction to the left external iliac artery. Carotid duplex showed dissection in the right common carotid artery extending to the internal and external carotid arteries. He then developed left sided-body weakness post-operatively. Case 2 was a 71 year old male with sudden onset of “tearing” chest pain radiating to the back. CT aortogram revealed Stanford A, Debakey I dissection from the aortic root extending to the right proximal common carotid artery down to the left mid external iliac artery. Carotid duplex showed dissection of right common carotid artery extending to the internal and external carotid arteries. He underwent emergency surgical repair and there were no neurologic deficits during admission. Case 3 was a 51 year old male with sudden onset of left leg weakness, numbness and poikilothermia. Acute limb ischemia was considered. Arterial duplex scan showed incidental finding of thrombus partially occluding the distal abdominal aorta and total occlusion of the left common iliac artery. CT aortogram revealed Stanford A, DeBakey I dissection with intraluminal thrombi in the right common carotid artery, and left common iliac to the external iliac arteries. Carotid duplex showed total occlusion of the proximal internal carotid artery secondary to a false lumen thrombus. He then developed right lateral preferential gaze prior to intervention.
Conclusions: Carotid artery dissection is an ominous complication that may result to permanent disabling cerebral malperfusion symptoms. This warrants an expeditious diagnosis for prompt medical and/or surgical management.
Duplex sonography of the right common carotid artery demonstrated different flow profiles. The false lumen showed systolic orthograde and retrograde diastolic flow, with decreased peak systolic velocity (comparing to the true lumen) while the true lumen is narrowed to >50% luminal stenosis.
Poster No. 90
Abstract ID No. 1498177
Triple threat: the terrible combination of bicuspid aortic valve, aortopathy and pseudocoarctation of the aorta in a patient with Turner syndrome
Nyssa Elline Palileo, MD, FPCP, FPCC, April Valdecañas, MD, FPCP, FPCC, Jenny Beltran, MD, FPCP, FPCC, FSVM, Shanon Marie Corsino, MD
St. Luke’s Medical Center - Quezon City
Background: Turner syndrome is a genetic condition in women associated with either complete or partial loss of one X chromosome. The most common congenital heart abnormalities are bicuspid aortic valve and aortic coarctation with prevalence rates of 15-30% and 7-18%, respectively. Aortopathy has been linked with Turner syndrome, possible mechanism due to TGF-beta signaling disruption. The incidence of Non-A Non B aortic dissection vary from 3-11% and challenges in treatment is well-recognised due to lack of of gold standard consensus.
Case presentation: A 23-year-old, female, Filipino diagnosed with 45,X karyotype at 3 year old (BSA of 1.5 m2) with bicuspid aortic valve and aortic root dilation of 2.1 cm/m2 by BSA. She is hypertensive and monitored by 2D echo doppler and MRA. At 23 year old, she had a sudden onset of severe “tearing” upper back pain. A long segment aortic dissection arising from the base of an aneurysmal left subclavian artery extending to the iliac bifurcation with a false lumen of 2.1 cm in the proximal third of the descending thoracic aorta was noted. There is also aortic root dilation and a mild to moderate short segment narrowing of the arch between the left common carotid and subclavian arteries. 2D echocardiogram noted bicuspid aortic valve with mild regurgitation and dilated aortic root. She was medically managed with losartan, carvedilol, statin and analgesics. Serial CT monitoring at 1 and 5 months showed a stable long segment thoracoabdominal aortic dissection, unchanged aneurysmal left subclavian artery and same degree of mild narrowing at the arch. Endovascular, hybrid and open surgical options were contemplated when there is recurrence of symptoms and findings such as increase in subclavian aneurysm size and aortic root size index to 2.5 cm/m2.
Conclusions: Turner syndrome with 45X karyotype, hypertension, bicuspid aortic valve and coarctation were at the highest risk to develop dissection. Non A non B aortic dissection has a complicated disease course; in addition to having genetic etiology with aortopathy, among the other risk factors of the case. Studies that demonstrate advantages of intervention, either open surgical and endovascular approach over medical management provide more satisfactory short term results.

Intimal flap arising from the base of the left subclavian artery take-off.
Poster No. 91
Abstract ID No. 1511593
Cyclist’s iliac syndrome
Sonal Pruthi, MD1, Sareena George, MD1, Sahil Parikh, MD2, Ari Mintz, DO1
1Columbia University Irving Medical Centre, 2Endovascular Interventional Cardiology Fellowship
Background: External iliac artery (EIA) endofibrosis is a poorly known, non-athereosclerotic cause of lower extremity pain in professional cyclists and runners. It is characterized by thickening of intima, accumulation of subendothelial connective tissue, collagen and smooth muscle with arterial lumen narrowing leading to claudication symptoms.
Case presentation: A 43-year-old female, avid runner (50 miles/week) and cyclist (intense cycling an hour a day), was evaluated for right lower extremity discomfort. She complained of a severe cramping sensation in her right calf with exercise which would subside with cessation in activity x 3 years and worsened to symptoms with minimal exertion and associated with numbness in her toes. Arterial ultrasound Doppler revealed occlusion of the right EIA with reconstitution of in the common femoral artery (CFA). Peripheral angiogram confirmed the findings of occluded EIA and reconstitution at CFA (Fig). An EKOS catheter was placed across the lesion and tPA (1mg/hour) infused for 12 hours. A re-look angiogram showed patent flow in the right EIA, and a non-flow limiting dissection extending from the EIA to CFA (Fig). IVUS confirmed the dissection, significant intimal and medial thickening consistent with endofibrosis (Fig). There was no significant disease otherwise. Decision was made to allow the CFA to heal and vascular surgical consultation for management of EIA endofibrosis. She was discharged on clopidogrel and Xarelto 2.5 mg BID and advised to abstain from strenuous physical activity.
Conclusions: External iliac artery endofibrosis is poorly known and symptoms are often misdiagnosed to be secondary to musculoskeletal or neurological causes. Drop in ankle brachial pressure index during exercise and ultrasound arterial dopplers are initial tests of choice. Treatment is mostly conservative, and includes avoiding strenuous exertion, and if it fails surgical intervention is the preferred definitive treatment. Complete occlusion of the artery with associated dissection as seen in our case is rare and though balloon angioplasty can be used as a temporary measure, only endovascular techniques is unsuitable.

Poster No. 92Withdrawn
Poster No. 93
Abstract ID No. 1500000
Unraveling the rarity: exploring the enigma of digital gangrene in a young female with vasculitis
Mark John D. Sabando, MD, Jan Tristan M. Acenas, MD, Bernadette Alexis M. Mariño, MD, Tam Adrian P. Aya-ay, N/A, MD, Paul Anthony O. Alad, MD, Roxanne Yen C. Bongcawil, MD, James Harold A. Barte, MD, Merrill Van C. Yu, MD, Muriel A. Morilla-Buco
University of the Philippines-Philippine General Hospital
Background: Systemic lupus erythematosus is an autoimmune disorder that affects the connective tissue and can cause vascular lesions among other multi-system manifestations. SLE-related vasculitis can take different forms, ranging from mild involvement of cutaneous vessels to severe catastrophic forms that lead to organ complications. Although less common, there may also be vascular wall necrosis and thrombus formation in the artery’s lumen. Digital ischemia resulting from SLE-associated vasculitis is rare.
Case presentation: A 26-year-old female with SLE presented with excessive vaginal bleeding and digital discoloration of both hands and feet. The patient had previously tested positive for ANA, anti-dsDNA, and Antiphospholipid Antibody Syndrome, but had not had recent follow-up due to the COVID-19 pandemic. Upon arrival at the emergency room, she tested positive for COVID-19. Physical examination showed violaceous discoloration of all the fingers and toes bilaterally. She had elevated creatinine, low hemoglobin, deranged coagulation and low complement C3, suggesting SLE activity. The patient received methylprednisolone pulse therapy as well as supportive blood transfusion. The violaceous discoloration seen in the fingers and toes of the patient progressed into dry gangrene which eventually demarcated. An arterial duplex scan showed no significant stenosis in the upper and lower extremities. Despite being recommended for amputation, the patient refused. After a multidisciplinary consultation, it was decided that auto-amputation would be the best course of action. The patient was discharged with improved condition and is being followed-up in the outpatient clinic.
Conclusions: This case report presented a young female with SLE who presented with digital gangrene, and discussed the approach and management of vascular complications in patients with SLE. Autoamputation was offered as a course of management for patients with SLE vasculitis leading to digital ischemia, with emphasis on the need to manage the underlying autoimmune disease. The report also highlighted the importance of a multidisciplinary approach for better long-term outcomes.

Progression of the violaceous lesions of both upper and lower extremities into gangrene.
Poster No. 94
Abstract ID No. 1474045
Isolated giant popliteal artery aneurysm mimicking a venous compressive syndrome—a case report
Joel B. Santos1, Jenny Beltran, MD, FPCP, FPCC, FSVM2
1Chinese General Hospital, 2St. Luke’s Medical Center-Quezon City
Background: The incidence of popliteal and femoral artery aneurysm were 7.4 per 100, 000 males and 1.0 per 100, 000 females. They usually affect men aged more than 60 years old with atherosclerotic cardiovascular disease and occur in association with other large vessel aneurysm (abdominal aorta, and femoral). The incidence of coexistent abdominal aortic aneurysm is 40%.
Case presentation: A 66-years old female, hypertensive, dyslipedemic, non-diabetic complains of right leg discomfort and varicosities. Patient experienced one year of right leg discomfort, heaviness, and aching, aggravated by prolonged standing and relieved by ambulation, with progressive enlargement of previously noted right leg varicosities with hyperpigmentation. One month thereafter, she noted sensation of fullness behind the right knee. Physical examination showed palpable pulsatile mass in the right fossa with tortuous non-axial varicosities of accessory saphenous vein distribution surrounding its periphery with hyperpigmentation on the right gaiter area. Cardio-pulmonary and abdominal findings were unremarkable. Arterial duplex scan showed giant saccular aneurysm of the right proximal popliteal artery measuring 8.77x4.66cm with short and narrow neck measuring 0.91x0.72cm without thrombus. CT peripheral angiography of lower extremities and abdomen revealed giant aneurysm of the proximal right popliteal artery measuring 4.5x4.6x7.8cm (APxTxCC) and absence of abdominal aortic aneurysm, respectively. She underwent resection of the right popliteal artery aneurysm through medial approach with distal femoral artery to distal popliteal artery interposition using polytetra-fluoroethylene graft. Quantiferon TB Gold Plus and histopathology were unremarkable. On follow-up, there was marked reduction of symptoms. However, there was no significant change on the appearance of the right leg varicosities. Repeat arterial duplex scan showed widely patent stent graft. She is scheduled for varicose vein ligation of accessory great saphenous and small saphenous varicosities.
Conclusions: The authors reports the first local case of isolated giant popliteal artery aneurysm mimicking a venous compressive syndrome

Giant popliteal artery aneurysm intraoperatively (a and b) and revascularization with polytetrafluoroethylene (PTFE) graft (c)
Poster No. 95
Abstract ID No. 1494287
A rare case of G-CSF induced large vessel vasculitis on an autoinflammation background
Ecem Sevim, MD1, Peter Grayson, MD1, Yogen Kanthi, MD2
1National Institutes of Health - National Institutes of Arthritis and Musculoskeletal and Skin Disease, 2National Institutes of Health
Background: Large vessel vasculitis (LVV) manifests as inflammation of the aorta and its major branches. Granulocyte colony-stimulating factor (G-CSF) can rarely cause LVV. Complex immune reactions may be responsible for the underlying etiology.
Case presentation: A 45-year-old male with a history of oral ulcers presented with abdominal pain and related symptoms, ultimately diagnosed with small intestine (jejunum) diffuse large B cell lymphoma and started on Rituximab, Etoposide, Prednisone, Vincristine, Cyclophosphamide and Doxorubicin regimen (R-EPOCH) with G-CSF. He developed recurrent bilateral carotidynia correlating with chemotherapy cycles. Imaging and laboratory studies did not reveal thrombosis or infection. CT scan of the neck revealed wall thickening in the common carotid arteries. PET-CT imaging demonstrated new, bilateral, intense arterial FDG uptake in the carotid, subclavian, aortic arch, descending aorta, and coronary arteries consistent with LVV. He was diagnosed with G-CSF-induced LVV and treated with oral prednisone between chemotherapy cycles, with symptom relief. Post-treatment PET-CT showed no FDG uptake in the neck and decreased uptake in the chest. The patient reported a history of recurrent oral ulcers since childhood and had a paternal family history of oral ulcers and various malignancies. Whole exome sequencing revealed a pathogenic truncating mutation in TNFAIP3. Loss-of-function mutations in TNFAIP3 have been associated with intestinal B cell lymphoma and can be a monogenic cause of A20 haploinsufficiency (HA20). HA20 is a newly described, monogenic autoinflammatory disease in which childhood-onset recurrent oral/gastrointestinal ulcers are a primary symptom. Vasculitis has been reported in patients with HA20. A20 is a potent inhibitor of the NFκB signaling pathway and associated pro-inflammatory cytokines. His inflammatory symptoms resolved after treatment with an IL-1 inhibitor, anakinra.
Conclusions: Treatment with G-CSF in rare instances can cause large vessel vasculitis. Underlying genetic risk factors, including mutations in TNFAIP3, may render specific patients susceptible to drug-induced forms of vasculitis.

Pre-treatment PET-CT (intense arterial FDG uptake in bilateral carotids)

Post-treatment PET-CT (no FDG uptake in carotids)
Poster No. 96
Abstract ID No. 1497339
Isolated elevated aPTT elevation in a patient with VTE- why does it matter?
Amitoj Singh, MD, Jessica Mintz, DO, Jana Montgomery, MD
Lahey Hospital and Medical Center
Background: Activated partial thromboplastin time (aPTT) is commonly used as part of coagulation studies and assesses both the intrinsic and common pathways of coagulation. Identifying the causes of elevated aPTT and its clinical significance particularly in the setting of VTE can help in making an earlier diagnosis and guiding management.
Case presentation: A 62-year-old female with past history of AML in remission status post stem cell transplant presented with acute onset left leg swelling and pain with phlegmasic symptoms with ambulation. There were no inciting events. She had an elevated aPTT level at 57s (normal 25-38s) without exposure to anticoagulation. Vascular ultrasound showed extensive, occlusive femoral-popliteal DVT. She remained symptomatic despite anticoagulation with enoxaparin. Invasive venography showed complete occlusion to the origin of left common iliac vein for which she underwent mechanical thrombectomy. She was discharged on apixaban; however, her labs returned post-discharge with positive lupus anticoagulant (LA), beta-2 glycoprotein and anti-cardiolipin antibody tentatively diagnosing her with APS. She was transitioned to warfarin for indefinite anticoagulation.
Conclusions: aPTT measures the activity of intrinsic and common pathways of coagulation. The differential of elevated aPTT includes APS, liver disease, DIC, and factor deficiencies. LA in cases of APS can falsely elevate PTT as it interferes with phospholipids used to induce in vitro coagulation. It is important to note that LA actually causes a prothrombotic effect in vivo, resulting in recurrent thrombosis that characterizes APS. Patients with presence of all three antibodies or “triple positive” have a high rate of recurrent thrombosis in APS with warfarin being the preferred choice of anticoagulation. APS is a rare disorder and the diagnosis typically involves a combination of clinical (vascular thrombosis) and laboratory criteria (antibodies). This case highlights having a high index of suspicion for APS in cases of isolated aPTT elevation and extensive clot as early recognition will ensure that the correct anticoagulant is chosen, hopefully preventing further thrombosis.
Poster No. 97
Abstract ID No. 1497491
Saccular femoral vein aneurysm as a nidus for deep vein thrombosis
Alexander E. Sullivan, MD1, Layne Janda, MD1, Tara Holder, MD1, Joshua Beckman, MD, MSc2, Daniel Clair, MD1
1Vanderbilt University Medical Center, 2University of Texas Southwestern Medical Center
Background: Femoral vein aneurysm is a rare vascular abnormality that may provide the nidus for deep venous thrombosis (DVT).
Case presentation: A 61-year-old man with a history of triple-positive antiphospholipid syndrome (APLS) and recurrent right popliteal DVTs presented for evaluation of chronic lower extremity edema and pain. He was initially diagnosed with a right lower extremity DVT five years prior and had a recurrent DVT four years later in the setting of a subtherapeutic INR despite taking warfarin. He since developed progressive, lifestyle-limiting right lower extremity edema, heaviness, and pain. Venous duplex ultrasonography showed nonocclusive echogenic thrombosis within the popliteal vein (panel A) extending proximally into the distal femoral vein (panel B) with reflux in the mid-femoral vein. He was referred for intervention to improve venous outflow and address symptoms. Initial venography demonstrated total occlusion of the popliteal vein (panel C). Multiple venoplasty attempts of the popliteal and posterior tibial veins were unsuccessful. After 24 hours of ultrasound-assisted thrombolysis, repeat venography demonstrated limited popliteal vein flow. Popliteal vein blood flow improved after successful popliteal vein and posterior tibial venoplasty. Venography at the conclusion of the case revealed a saccular distal femoral vein aneurysm (panel D). The aneurysm was felt to be the nidus for recurrent DVT in the setting of APLS. The patient was continued on compression therapy and therapeutic warfarin. Post-thrombotic symptoms were improved at 1-month follow-up.
Conclusions: We present a case of a patient with established APLS with subsequent DVT in the setting of an idiopathic femoral vein aneurysm. Despite underlying thrombotic disorders, contemplation of a “two-hit hypothesis” with a second risk factor tipping the bleeding-clotting homeostatic balance may reveal a treatable condition.

Poster No. 98
Abstract ID No. 1511425
Bilateral renal artery aneurysm and infarction in the setting of COVID-19: an epiphenomenon or fibromuscular dysplasia in disguise?
Sneha E. Thomas, M.D.1, Raghu Vallabhaneni, M.D.2, Elizabeth Ratchford, MD3
1University of Maryland Medical Center-Midtown, 2MedStar Health, 3Johns Hopkins School of Medicine
Background: Severe acute respiratory syndrome coronavirus 2 infection (COVID-19) has recently been found to target the kidneys. However, only a handful of cases have reported renal artery thrombosis or aneurysm.
Case presentation: A 40-year-old female with no known atherosclerotic vascular disease presented with sudden onset of right flank pain. She tested positive for COVID-19 five days before and had mild symptoms. Imaging showed a hypodense wedge-shaped focus in the upper pole of the right kidney. She was discharged home on anticoagulation. The next day she returned with left-sided flank pain and had a new left renal infarction. CTA abdomen/pelvis showed wall thickening with irregularity in the periphery of the right upper renal artery and irregularity of the mid left renal artery. TTE was normal and event monitoring showed no arrythmias. Hypercoagulable workup noted positive lupus anticoagulant and mildly positive anticardiolipin antibodies. Enoxaparin was started and later transitioned to warfarin. Her vascular surgeon felt the imaging was consistent with fibromuscular dysplasia (FMD) and was referred to our FMD Clinic. Serial follow-up CTAs with 3-D-reconstruction revealed persistent beading of the renal arteries and an 8 mm aneurysm in the uppermost right renal artery (Figures 1a-1b). CTA head/neck showed tortuosity of the bilateral internal carotid arteries without frank beading or aneurysm.
Conclusions: This unique case illustrates bilateral renal artery infarction in a 40-year-old female with mild COVID-19 infection. Further advanced imaging including 3-D-reconstruction was required to differentiate renal artery irregularities on imaging were secondary to viral inflammatory response in COVID-19 versus multifocal FMD. In rare occasions, underlying vascular abnormalities may predispose the renal arteries to injury. Transient antiphospholipid antibodies may also have played a role, which has been reported in the context of COVID-19. Close clinical follow up with surveillance imaging is necessary in these ambiguous clinical situations to rule out an underlying vasculopathy.

Poster No. 99
Abstract ID No. 1498657
Concurrent antiphospholipid syndrome with recurrent pulmonary embolism in a young female with takayasu’s arteritis: a case report
April Valdecañas, MD, FPCP, FPCC, Lester Veroy, MD, FPCP, FPCC, FPSVM
St. Luke’s Medical Center Quezon City
Background: The association between Takayasu’s arteritis (TA) and presence of antiphospholipid antibody has been reported, however, TA with concurrent antiphospholipid syndrome (APS) is rare.
Case presentation: A case of a 34-year old female, previously diagnosed with TA with pulmonary artery involvement and APS, presented with chest pain and dyspnea. Focused physical examination showed clear lung fields and weak pulses on left brachial, radial, and ulnar arteries. Repeat computed tomography pulmonary angiogram revealed the same degree of severe narrowing of the distal right main pulmonary artery and occlusive changes in the ipsilateral pulmonary arteries; also, with the same degree of secondary collateral vessel formation, left pulmonary artery dilatation, as well as no change in occlusive changes in the left lower lobe segmental branches. However, a new filling defect was noted in the periphery of superior segmental pulmonary artery ascribed as embolism, which was managed as a submassive pulmonary embolism. Treatment-changing factors include patient co-morbidities and VTE recurrence.
Conclusions: This is a rare case of concurrent TA and APS complicated by recurrent pulmonary embolism. Physicians should bear in mind that multiple etiologies may exist in a single patient despite the rarity. Managing complicated cases require timely and careful considerations of suitable intervention/s. A multidisciplinary approach is recommended to help weigh the risks and benefits of each possible treatment. Further studies of TA and APS regarding clinical and prognostic differences are needed.

CT pulmonary angiogram depicting filling defect in the right superior segmental pulmonary artery.
Poster No. 100
Abstract ID No. 1498597
Acute partial aortic occlusion presenting as transient Adamkiewicz syndrome in a 68-year-old female with acute decompensated heart failure: a case report
Patrick N. Vera Cruz, MD, FPCP, Nyssa Elline Palileo, MD, FPCP, FPCC, Angelo Panelo, MD, DPCP, Lea Arceli Gonzales Porciuncula, Castro T. Rod, MD, FPCP, FPCC
St. Luke’s Medical Center - Quezon City
Background: The artery of Adamkiewicz is a unilateral artery arising from the left side of the aorta between the T11 to L2. Obstruction of this artery leads to a syndrome characterized by sudden, painless lower extremity weakness, variable sensory deficit, and relative sparing of proprioception. Here, we present a case of a transient Adamkiewicz syndrome secondary to a thromboembolic acute partial aortic occlusion posing unique diagnostic and management dilemmas.
Case presentation: We are presented with a 68-year-old female with known heart failure, atrial fibrillation, and history of cardioembolic stroke coming in for shortness of breath and fever. She was initially managed as a case of acute decompensated heart failure, community acquired pneumonia, and hypertensive emergency hence antibiotics, antihypertensives, oral anticoagulation, and heart failure medications were given. On the first hospital day, she suddenly developed numbness and lower extremity paralysis. Examination showed complete motor and sensory loss extending distally from the T11 dermatome, areflexia, with relative sparing of proprioception. Craniospinal MRI showed a small hyperacute infarct at the left frontal lobe and absence of any spinal lesion. A few hours later, gradual improvement was noted with complete recovery 21 hours post-ictus. However, her legs became pulseless with absent doppler signals at the bilateral dorsalis pedis and posterior tibial arteries. Unfractionated Heparin was then started to maintain an INR of >2.0. CT aortogram showed multiple thrombi at the left atrium and the infrarenal abdominal aorta from L2 to L4. Arterial duplex scan showed partial aortic occlusive disease, atherosclerotic with acute thromboembolic component. Anticoagulation was continued and there was gradual improvement of circulation and overall clinical status precluding the need for surgical intervention until discharge.
Conclusions: Firstly, this case emphasizes that heart failure patients have an increased risk for thromboembolic events. Secondly, thorough examination is crucial to localize the ischemic lesion. Finally, conservative management for acute partial aortic occlusion is a viable option for high surgical risk patients who are clinically stable.
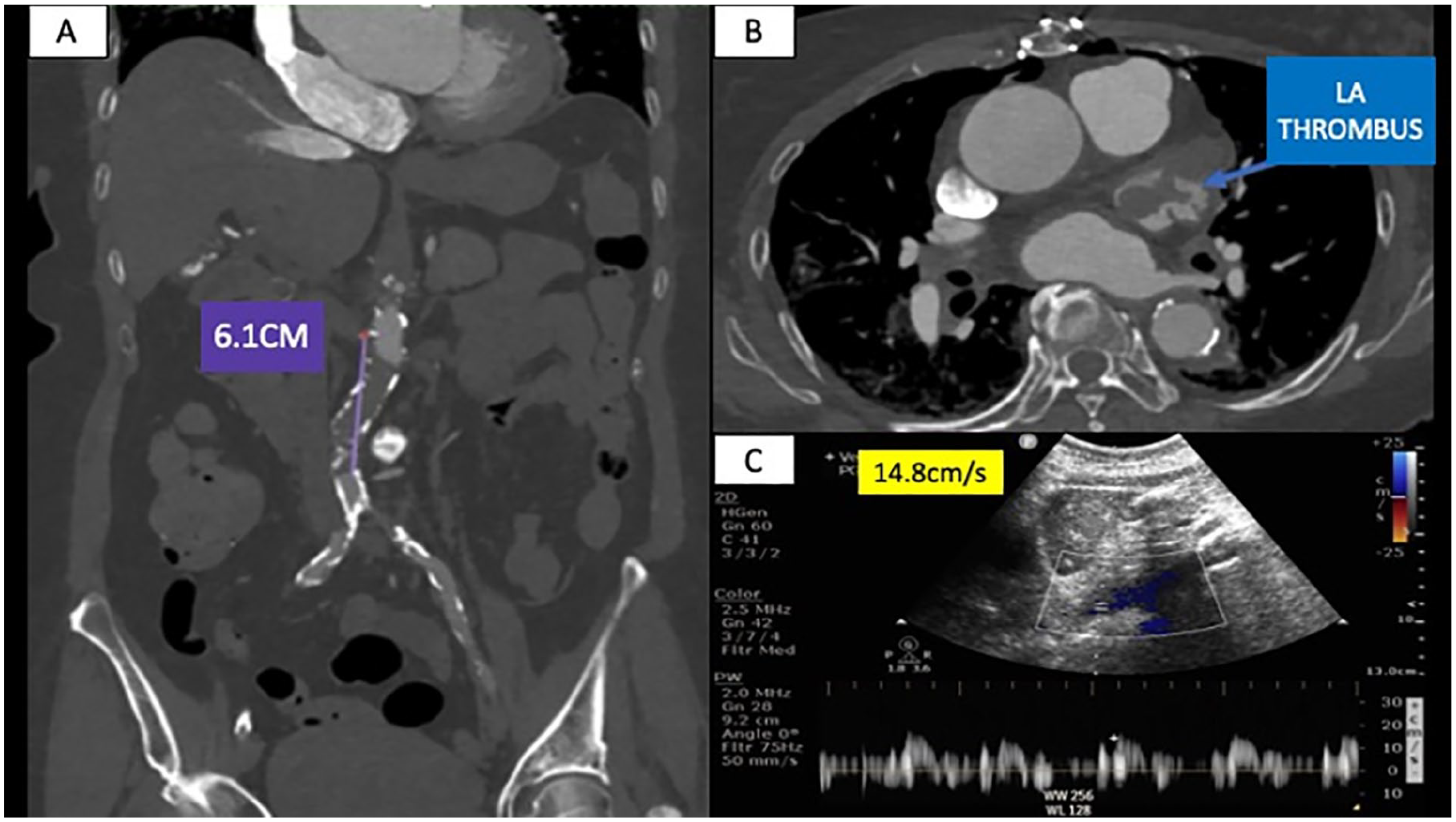
Multiple thrombi at the infrarenal aorta measuring 6.1cm in length (A) and the left atrial appendage (B). Distal abdominal aorta showing partial obstruction with peak systolic velocity of 14.8cm/s.
Poster No. 101
Abstract ID No. 1486150
A 55-year old Filipino female with Klippel-Trenaunay syndrome: a case report
Aaron Christian, Earl Vidad
Philippine General Hospital
Background: Klippel Trenaunay syndrome (KTS) is a complex congenital vascular syndrome characterized by a triad of (1) extensive nevus of the affected limb, (2) venous malformations on the involved limb, and (3) hypertrophy of all tissue, especially of the bones in the impaired limb. Because KTS is a rare congenital syndrome with varied clinical manifestations, early recognition, prevention, and treatment of complications are paramount. KTS deserves attention through this report since there is no Philippine registry about this rare condition.
Case presentation: We report a case of a 55-year-old Filipino female who has had a known case of schizophrenia since 2019 but is non-compliant with medications and was lost to follow-up. She was referred to cardiology service for evaluation of enlarging left lower extremity. Further history revealed, she was allegedly born with unequal limb girth of bilateral lower extremities, with prominent tortuous soft masses on the left lower extremity since childhood. Six years before admission, she noted darkening of the skin on the anterior left lower leg, with spontaneous vesicle eruptions followed by drying and complete healing—no further consultation since. Workup was done, including MRI of the left lower extremity, revealed multiple dilated vessels in the subcutaneous and intramuscular layers suggestive of varicose veins with associated phleboliths. These findings are compatible with KTS.
Conclusions: KTS presents varied clinical manifestations, mostly during childhood. A high clinical index of suspicion and imaging has been at the forefront in diagnosing this rare complex vascular syndrome. Magnetic resonance imaging with short tau inversion recovery sequence is one of the non-invasive imaging modalities for this disorder.

Physical examination of the left lower extremity: Patchy areas of violaceous discoloration on the anterolateral aspect of the thigh; tortuous prominence, soft, non-tender on palpation, diffused on left thigh and Leg. Dark skin discoloration on distal anterior leg with superficial ulcerations.
Poster No. 102
Abstract ID No. 1486091
Antiphospholipid syndrome (Hughes syndrome) mimicking vasculitis in a 24-year-old female: a case report
University of the Philippines- Philippine General Hospital
Background: Antiphospholipid syndrome (APS) is an autoimmune disease presenting hypercoagulability with variable clinical presentation. These include vascular thrombosis and obstetric complications with repeated miscarriages among women. We report a young Filipino female with features mimicking vasculitis, no typical clinical presentation of antiphospholipid syndrome, and no history of trauma, oral contraceptive pills, or recent surgery. Moreover, laboratory examinations revealed positive for ApL antibodies.
Case presentation: A 24-year-old Filipino female admitted for chest pain and decreased left upper extremity sensation. She has no other comorbidities, no prior contraceptive use, or vices. She had two episodes of miscarriages at 16th week of gestation last 2021. One month before admission, she had intermittent chest pain varying in character from squeezing to pricking with moderate to severe intensity with associated peripheral cyanosis and decreased sensation in her left upper extremity, which lasted for about one week. Three weeks before admission, persistence of symptoms prompted consult with a cardiologist, with primary consideration of a possible vasculitis hence the following workup: arterial duplex scan of both upper extremities revealed a subacute to chronic peripheral arterial insufficiency with 60 to 70% stenosis of the distal subclavian and axillary arteries; with persistently high resistive indices could be secondary to vasculitis among other. Baseline TTE revealed unremarkable findings. She was given Prednisone 10mg/tab, one tablet daily. Interim, there was temporary relief of symptoms until one hour before admission (May 2022); noted stiffening and numbness of upper extremities and inability to move for about an hour with no associated slurred speech or sensorimotor deficits. This was associated with nape pain and tinnitus.
Conclusions: A diagnosis of the antiphospholipid syndrome remains a challenge among clinicians due to an expanding range of clinical manifestations not included in the current classification criteria associated with the presence of antiphospholipid antibodies. Despite our knowledge of this disease and the pathogenic mechanisms involved, many remains must be done.

Pertinent laboratory test results.

Arterial duplex scan of left upper extremity showing persistently high resistive indices of the distal subclavian and axillary arteries with about 60 to 70% stenosis.
Poster No. 103
Abstract ID No. 1510161
Coronary artery aneurysm and systemic vasculopathy
Hai Xu, MD1,2, Jessica K. Bjorklund, MD, MPH1
1Staten Island University Hospital / 2Northwell Health
Background: Characterization of the coronary artery aneurysm (CAA) pathophysiology and its underlying association with systemic vasculopathy is critical to decision making in therapeutic interventions. We present a case of RCA aneurysm in an adult patient to highlight the implications of multimodality imaging in guiding diagnosis as well as key points in management of associated aortopathy.
Case presentation: A 59-year-old male with past medical history of hyperlipidemia and hypertension presented for dyspnea on exertion. Otherwise, patient denied relevant social or family histories. Transthoracic echocardiogram demonstrates normal systolic and diastolic functions with ascending aorta dilatation at 4.4cm. Coronary CTA showed predominantly thrombosed fusiform aneurysm involving the entire RCA measuring up to 3.6cm. CT-FFR confirmed there is no hemodynamically significant lesions. Patient was subsequently optimized on medical therapy and referred to cardio-thoracic surgery for further evaluation. Genetic counselling was also initiated to explore underlying association among multiple manifestations of vasculopathy in this patient.
Conclusions: Despite anecdotal evidence, no prospective data has supported the use of chronic anticoagulation in this rare entity. Coronary artery bypass grafting has been used for the treatment of co-existing significant obstructive CAD. Non-invasive multi-modality imaging provides comprehensive and complementary information for CAA as well as its associated systemic vasculopathy. Based on published literature, incidences of CAA in patients with ascending aortic aneurysms is 17%. Congenital or acquired connective tissues/inflammatory diseases affecting the aorta and coronaries maybe the underlying mechanism and should be considered as a continuum. Further evaluation for underlying genetic disorders is crucial in management strategies.

Thrombosed fusiform aneurysm involving the entire right coronary artery measuring up to 3.6 cm.
Poster No. 104
Abstract ID No. 1498421
A case of acute limb ischemia in a patient with Pseudomonas aeruginosa septicemia
Julianne Marie E. Yamamoto, Ramayana G. Garcia, Lester S. Uy, Bernard M. Baluga, Jose Maria Mendieta, Justine P. Cabrera
St. Luke’s Medical Center - Bonfacio Global City
Background: Acute limb ischemia (ALI) is a dreaded vascular emergency that can lead to loss of limb or life. Traditional risk factors include cardioembolic, atherothrombotic, and coagulopathy. Infection as a cause to trigger ALI is at times overlooked.
Case presentation: A 51-year old male, known hypertensive and dyslipidemic, initially admitted as a case of septicemia of Pseudomonas aeruginosa secondary to acute tonsillopharyngitis, presented with sudden onset of severe bilateral lower leg pain. Arterial duplex scan showed total occlusion of the distal arteries of the bilateral lower extremities with acute thrombus in the right popliteal artery and monophasic waveform pattern in the proximal arterial segments. CT aortogram showed long-segment filling defect in the infrarenal segment of the abdominal aorta, bilateral common iliac, internal iliac, and external iliac arteries suggesting aorto-iliac occlusive disease, with an incidental finding of portal vein thrombosis. Intravenous anticoagulation and emergency embolectomy of the arterial segments of the bilateral lower extremities were done. Full recovery was achieved. Patient was discharged on oral anticoagulation.
Conclusions: Patients without traditional risk factors for ALI such as cardiac arrhythmia, atheromatous plaques, coagulopathies and iatrogenic causes should be investigated for other non-traditional causes. Severe infection as a cause to develop acute arterial and venous thrombosis should always be considered.
Arterial thrombus from the right lower extremity.
Poster No. 105
Abstract ID No. 1496248
Two strokes of luck: a case of descending thoracic aneurysm in an elderly patient with multiple aortopathies and a previous abdominal aortic aneurysm repair
Julianne Marie E. Yamamoto, Flordeliz F. Lontok, Ronald S. Reodica, Fabio Enrique B. Posas, Marvin Martinez, Precious Samonte, Arvinjay Bautista
St. Luke’s Medical Center - Bonfacio Global City
Background: Treating a patient with aneurysms in multiple segments of the aorta and its branches is a challenge, especially in the elderly with a history of open surgical repair of abdominal aorta aneurysm. Management including staged interventions is important in multiple aortopathies.
Case presentation: A 78-year old male with previous abdominal aortic aneurysm surgical repair 12 years ago, with history of chronic cough, was admitted for chest CT scan evaluation of a lung mass seen on chest x-ray. The patient is hypertensive, previous smoker, with chronic kidney disease. CT scan result showed a saccular aneurysm in the descending aorta located 1.8cm after the take-off of the left subclavian artery. Fusiform aneurysm in the right brachiocephalic area, chronic thoracoabdominal aortic dissection, and small saccular outpouchings in the infrarenal abdominal aorta were also noted. The infrarenal bypass graft from the previous aortic repair was patent. Cervical debranching with Y-graft anastomosis of the brachiocephalic artery to the left common carotid and left subclavian arteries was done, followed by thoracic endovascular repair of the saccular aneurysm. Both procedures were uneventful. Surveillance and treatment of the other aortopathies are underway.
Conclusions: A step-wise approach treatment is key in multiple aortopathies, including the decision of which diseased segments must be addressed first, the optimal timing to intervene, and plan the succeeding management to have better outcomes in these patients.

CT Aortogram showing multiple diseased areas of the aorta.