
Abstract
Keywords
Introduction
The Ehlers–Danlos syndromes (EDS) are a heterogeneous group of hereditary connective tissue disorders affecting collagen production, structure, and function, characterized by a range of clinical features, from joint hypermobility and tissue fragility to arterial aneurysms, dissections, and organ rupture. 1 Recently, updated nosology delineates 13 EDS subtypes, highlighting associated co-morbid conditions. Within these guidelines, the commonest form, hypermobile EDS (hEDS), is further distinguished from the related hypermobility spectrum disorders (HSD), but with a higher expected incidence of systemic complications. 2
Potentially serious cardiovascular manifestations are well cited in the vascular, classical, and cardiac-valvular forms of EDS.3–5 In contrast, only mild, nonprogressive thoracic aortic dilatation and mitral valve prolapse (MVP) have historically been recognized as cardiovascular features of hEDS.6,7 Incidence of these disorders were previously reported between 6% and 21%,8,9 prompting echocardiography to be routinely included as part of the evaluation of joint hypermobility syndromes. Although these initial reports were based on small cohorts with mixed ages that included other forms of EDS and predated the updated nosology, both aortic root dilatation and MVP are included as part of the 2017 hEDS diagnostic criteria. 2 In the contemporary diagnostic era, echocardiography is performed in up to 90% of patients under evaluation for EDS 10 but, at least in children and young adults,10–12 has not been found to reveal significant cardiac pathology, 11 prompting some to suggest that echocardiographic screening may not be necessary as part of a routine hEDS evaluation. Additionally, extra-thoracic arterial complications, such as spontaneous cervical artery dissection (CeAD),13,14 have been rarely reported prior to 2017 in patients with hEDS as part of broader vascular disease series. Notably, a recent comprehensive review of vascular phenotypes in nonvascular subtypes of EDS did not include hEDS or HSDs. 4 The incidence and range of severity of cardiovascular complications in older adults with hEDS or HSD have not been systematically evaluated in the era of the updated 2017 nosology.
Methods
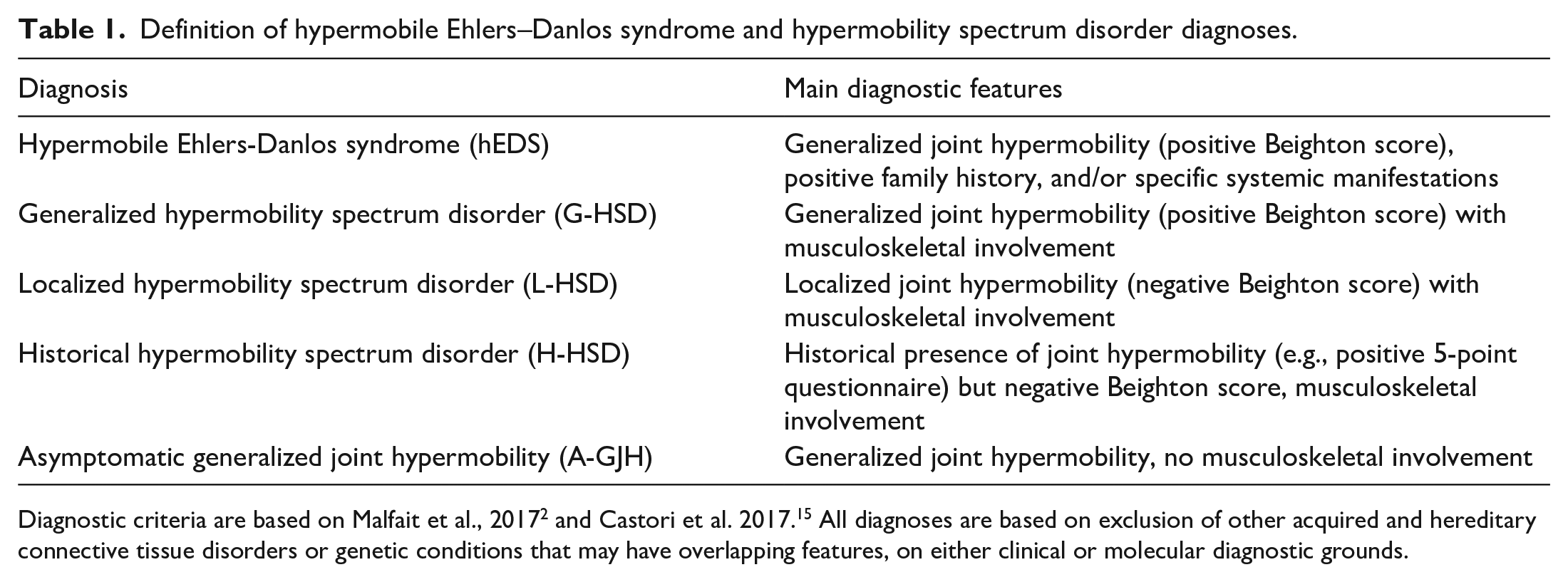
The study population included patients referred to our multidisciplinary Cardiovascular Genetics Program for evaluation of a heritable connective tissue disorder between January 2017 and December 2020 for suspected joint hypermobility-related symptoms by cardiologists, primary care providers, rheumatologists, orthopedists, pain management specialists, neurologists, vascular medicine specialists or self-referred, and evaluated by a study investigator (ARK, BDG or LM). Electronic health records (EHR) were reviewed for demographic data, diagnoses of hEDS or HSD (based on updated 2017 criteria2,15; see Table 1 for definitions) documented by a study investigator, other cardiovascular diagnoses, and clinical genetic testing results where available. The study was conducted under the approval of Mount Sinai’s Institutional Review Board (GCO # 19-0883 ISMMS) utilizing a waiver of informed consent.
Definition of hypermobile Ehlers–Danlos syndrome and hypermobility spectrum disorder diagnoses.
Echocardiographic reports were reviewed for all subjects where available and data were systematically collected, including presence or absence of MVP, mitral valve insufficiency presence/severity, and end-diastolic aortic diameters at the sinuses of Valsalva, tubular ascending segment and arch measured from parasternal long-axis views perpendicular to the long axis of the aorta, using the leading edge technique. Ascertainment of aortic dilatation was determined by evaluating the documented maximal thoracic aortic diameters within established aortic nomograms for age, sex, and height 16 ; measurements ⩾ 2 SDs above the mean were deemed dilated.
Descriptive statistics for categorical variables were reported as frequency and percentages. Statistical analysis was performed using Microsoft Excel. Fisher’s exact test (two-tailed) was used to compare rates of aortic dilatation between sexes and in hEDS versus alternative hypermobility diagnosis (HSD or hypermobility disorder not otherwise specified). A p ⩽ 0.05 was considered statistically significant.
Results
During the study period, 258 patients met the inclusion criteria and were diagnosed with either hEDS or an HSD (Table 1). The median age was 31 years (range 4–73; 94.2% ⩾ 18 years) and 90% were female. The diagnosis of hEDS was made in 116 (45%) subjects (Table 2).
Subject demographics.
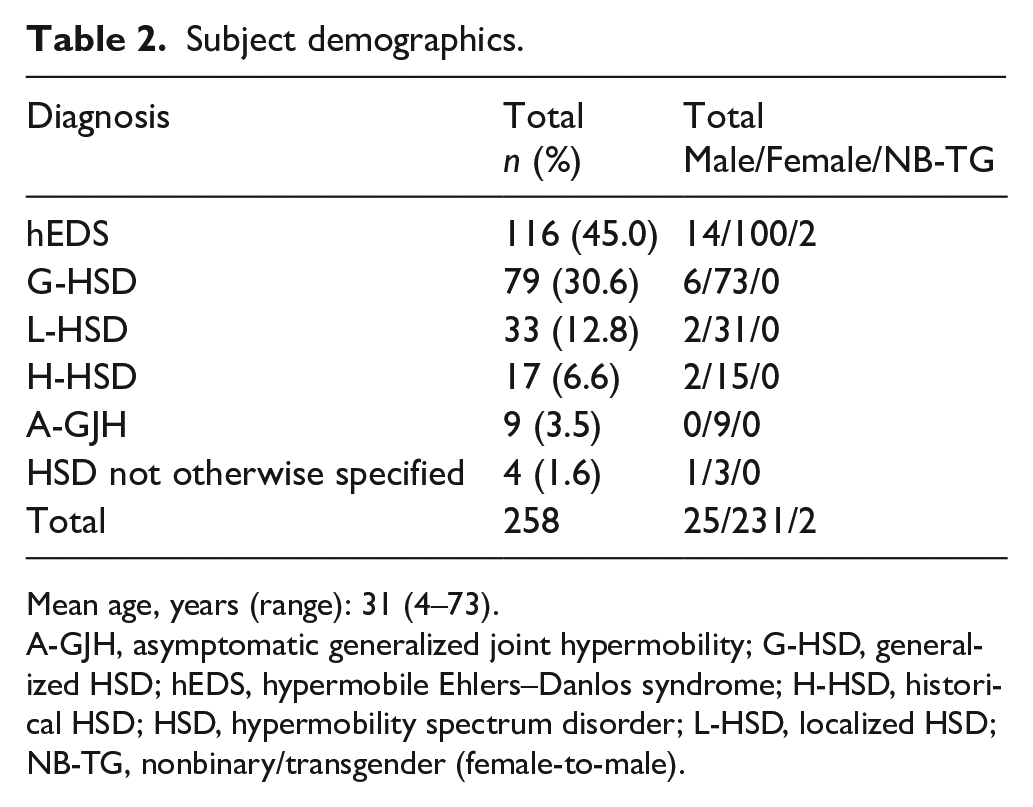
Mean age, years (range): 31 (4–73).
A-GJH, asymptomatic generalized joint hypermobility; G-HSD, generalized HSD; hEDS, hypermobile Ehlers–Danlos syndrome; H-HSD, historical HSD; HSD, hypermobility spectrum disorder; L-HSD, localized HSD; NB-TG, nonbinary/transgender (female-to-male).
Valvular assessment
Of the 254 subjects (98.4%) with echocardiographic data available, 19 (7.5%) had MVP and five (2.0%) had a structural mitral valve abnormality such as borderline prolapse, bowing or buckling. The incidence of MVP was similar in individuals with hEDS and HSDs (8, 42% vs 11, 58%, p = 0.17). Mild mitral regurgitation was seen in 14 individuals and mild-to-moderate in two (Table 3).
Mitral valve anomalies and severity in 258 subjects with hypermobile Ehlers–Danlos syndrome or hypermobility spectrum disorder with available echocardiograms.

Thoracic aortic assessment
Among 230 echocardiograms reporting aortic dimensions, the thoracic aorta was dilated in 35 (15.2%), upper limit of normal in 12 (5.2%), and normal in 183 (79.6%) individuals. The prevalence of aortic dilatation was higher in patients diagnosed with hEDS (24/116, 20.7%) compared to the pooled HSD cohort (11/142, 7.7%) (p = 0.003). There were similar proportions of males (4/25, 16.0%) and females (31/231, 13.4%) with hEDS/HSD who had aortic dilatation (p = 0.76). However, males with dilatation had higher maximal thoracic aortic diameters than females (mean 4.3 vs 3.5 cm, p < 0.001), despite the male subcohort being slightly younger (median age 33.5 vs 44 years, p = 0.31) and of similar body surface area (BSA) (1.87 vs 1.78 mm2, p = 0.052). Among all individuals with hEDS/HSD, there was a trend in increasing prevalence of aortic dilatation by age (Figure 1). Dilatation was mild in > 90% of females and did not show dramatic rise by decade of life (Table 4). However, two of the four males had moderate-to-severe dilatation (4.6 cm and 4.9 cm), which was confirmed by magnetic resonance angiography.
Age distribution of hypermobile Ehlers–Danlos syndrome or hypermobility spectrum disorder diagnoses and within-group average maximal thoracic aortic diameters among females and males with noted aortic dilatation.
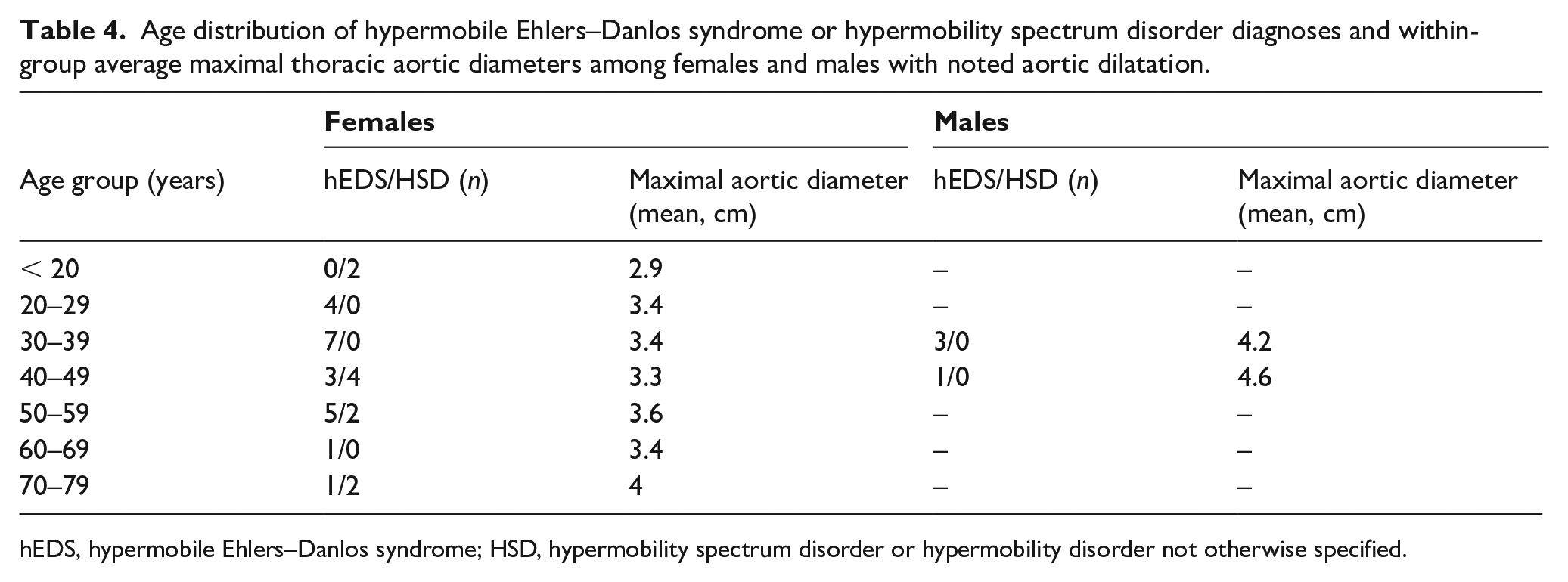
hEDS, hypermobile Ehlers–Danlos syndrome; HSD, hypermobility spectrum disorder or hypermobility disorder not otherwise specified.
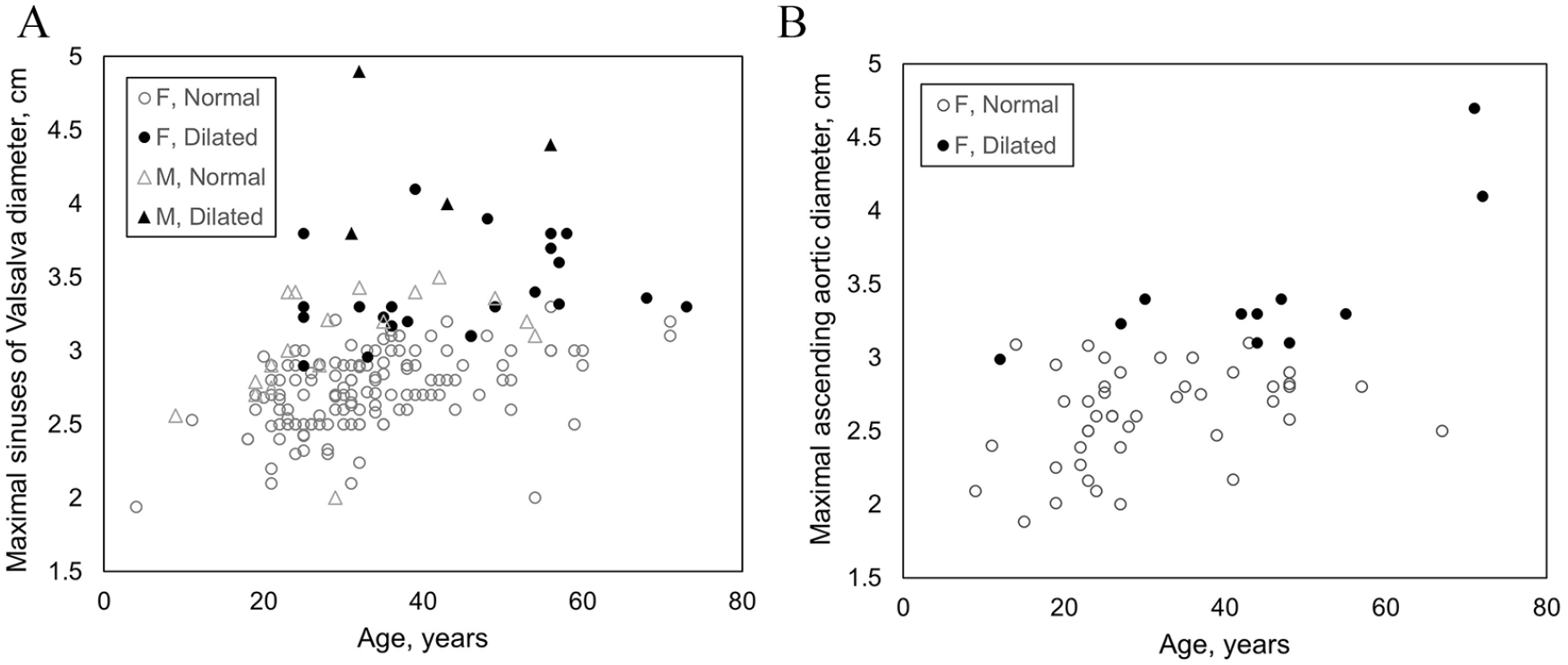
Distribution of thoracic aortic diameters by age measured in individuals with hypermobile Ehlers–Danlos syndrome or hypermobility spectrum disorder with available echocardiographic data. Measurements are represented according to anatomic location where aortic diameter was maximal, either (
Extra-aortic arterial features
Extra-aortic arterial involvement was present in five patients, four of whom also had aortic dilatation (Table 5). Two, both also with aortic root dilatation, had CeAD: one (hEDS) had bilateral spontaneous carotid artery dissection and the other (H-HSD) had dissection of the distal left internal carotid artery with a dissecting pseudoaneurysm of the left vertebral artery. Three individuals had a diagnosis of spontaneous coronary artery dissection (SCAD) or angiographic evidence of this confirmed by a study investigator (DKD or JWO); two of these (both female) had aortic root dilatation and one (male, G-HSD) did not. One female with hEDS and both SCAD and aortic root dilatation also had a focal outpouching of the proximal celiac artery (5 mm width × 5 mm depth) suggestive of a saccular aneurysm. All five individuals were assessed and ruled out for fibromuscular dysplasia (FMD) by a study investigator (DKD or JWO). Extra-aortic findings were similarly distributed between cases of hEDS (n = 2) and HSDs (n = 3).
Arterial phenotypes in five subjects with hypermobile Ehlers–Danlos syndrome or hypermobility spectrum disorder.

CeAD, cervical artery dissection; G-HSD, generalized hypermobility spectrum disorder; hEDS, hypermobile Ehlers–Danlos syndrome; H-HSD, historical hypermobility spectrum disorder; PA, pseudoaneurysm; SCAD, spontaneous coronary artery dissection.
Genetic assessment
Genetic testing through Clinical Laboratory Improvement Ammendments (CLIA)-approved commercial laboratories was performed as part of the clinical evaluation (indicated on the basis of either personal history of aortic dilatation or extra-aortic arterial findings or family history of aneurysm or dissection) or available from outside evaluations on 41/258 (15.9%) subjects. Targeted gene sequencing for vascular ± classical EDS (either COL3A1 alone or with COL5A1 and COL5A2) was performed in 16 individuals, and either a comprehensive aortopathy/arteriopathy gene panel (including the genes related to Marfan, FBN1; Loeys–Dietz, TGFBR1, TGFBR2, SMAD3, TGFB2; and vascular EDS, COL3A1 syndromes, along with additional nonsyndromic heritable thoracic aneurysm and dissection genes) or a heritable connective tissue disorders panel was used in 25 cases. Of 22 patients with aortic dilatation and five patients with extra-aortic manifestations who underwent genetic testing, results were either negative or identified a variant of uncertain significance (VUS) (Table 6). One additional subject, a 35-year-old female, had been referred after carrier screening during pregnancy revealed a likely pathogenic partial deletion in TNXB (121-bp deletion in exon 35); this subject was diagnosed with localized HSD (L-HSD) and had no cardiovascular phenotype. Heterozygous pathogenic variants in TNXB, which encodes tenascin-X, an extracellular protein that regulates elastic fiber integrity, have been described in case series as causing features of EDS (joint hypermobility, dislocations, and chronic pain). 17 Interestingly, this patient had multiple affected relatives with hypermobility symptoms, but none was found to carry the TNXB variant, suggesting that it may not have been causative or there may be an oligogenic etiology to her L-HSD. Two individuals, neither with cardiovascular phenotypes, carried pathogenic variants that were interpreted to be unassociated with their presentations. One was a 33-year-old female with hEDS who had prior genetic testing on a heritable connective tissue diseases panel and was found to be heterozygous for a pathogenic variant in FKBP14. Since this gene is associated with autosomal recessive kyphoscoliotic EDS, the result was interpreted to be clinically insignificant. Another individual, a 61-year-old female with L-HSD, proceeded to genetic evaluation for a possible metabolic condition and was found on whole exome sequencing to harbor a heterozygous pathogenic variant in FLG; this gene is associated with ichthyosis vulgaris and was not relevant to HSD. Following completion of genetic testing, no alternative genetic arteriopathy was apparent in the patients with arterial findings, other than clinically diagnosed hEDS/HSD.
Reportable genetic variants in subjects with hypermobile Ehlers–Danlos syndrome or hypermobility spectrum disorder.
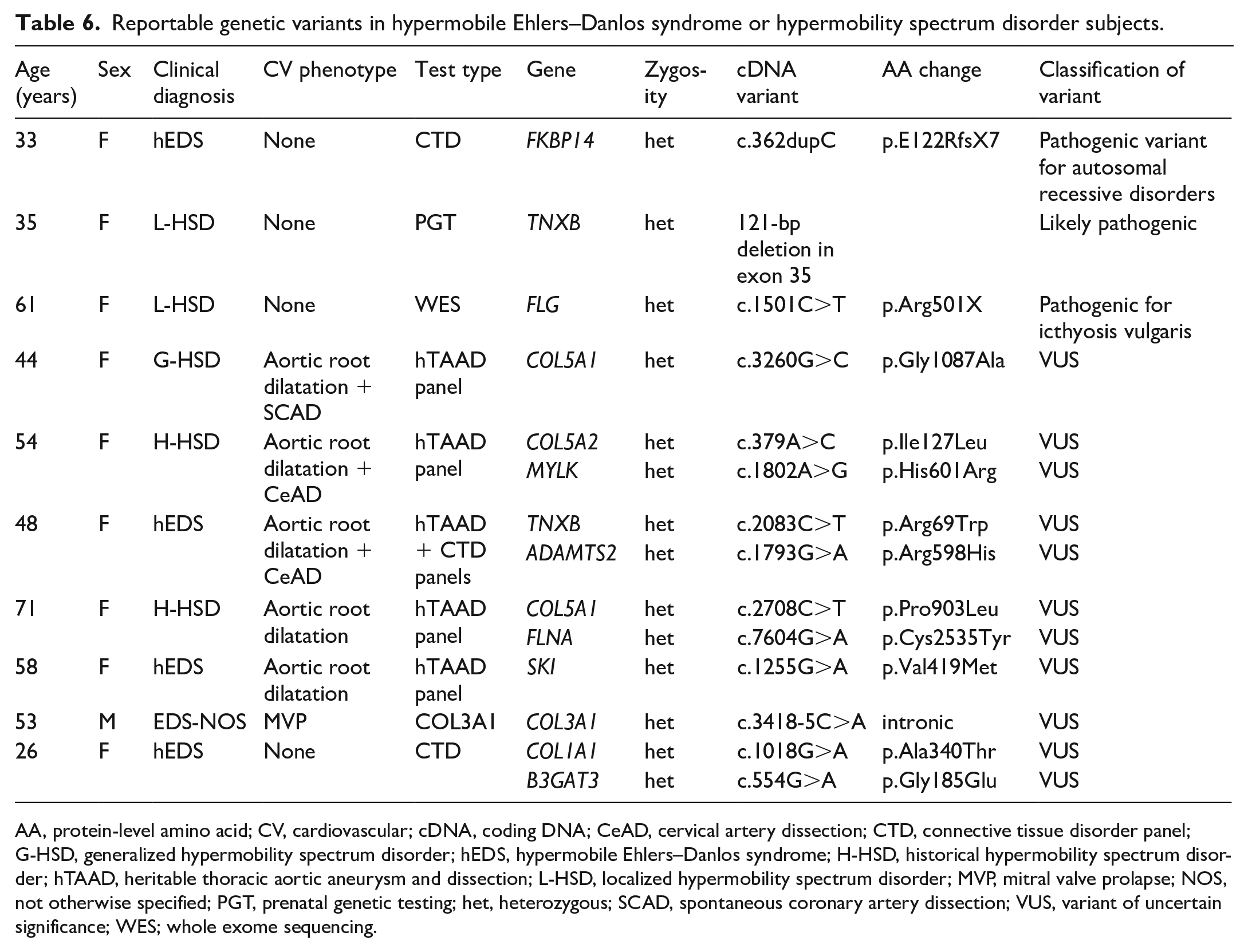
AA, protein-level amino acid; CV, cardiovascular; cDNA, coding DNA; CeAD, cervical artery dissection; CTD, connective tissue disorder panel; G-HSD, generalized hypermobility spectrum disorder; hEDS, hypermobile Ehlers–Danlos syndrome; H-HSD, historical hypermobility spectrum disorder; hTAAD, heritable thoracic aortic aneurysm and dissection; L-HSD, localized hypermobility spectrum disorder; MVP, mitral valve prolapse; NOS, not otherwise specified; PGT, prenatal genetic testing; het, heterozygous; SCAD, spontaneous coronary artery dissection; VUS, variant of uncertain significance; WES; whole exome sequencing.
Discussion
A contemporary description of the full spectrum of cardiovascular manifestations in patients with the commonest form of EDS, hEDS, and related HSDs across all age groups was previously lacking. We, therefore, retrospectively assessed 258 subjects with hEDS or HSD diagnosed according to the updated 2017 clinical criteria. The two cardiovascular conditions that have historically been cited in association with hypermobility disorders, MVP2,18 and aortic root dilatation, 6 were seen in 7.5% and 15.2% of the subjects with available echocardiograms, respectively, with severity typically mild. MVP was enriched compared to the expected population prevalence of 0.7%. 19 Since hEDS and HSD, as defined in the updated 2017 guidelines, occur on a spectrum and share many systemic associations, 20 it is not surprising that we found a similar incidence of MVP among both diagnoses.
Previous studies have documented an association between thoracic aortic dilation and joint hypermobility syndromes, but this is emerging as an age-dependent phenomenon. Although pre-2017 pediatric series cited the rate of aortic root dilatation in children with hEDS at 14%, 12 recent studies including children meeting the more stringent 2017 hEDS criteria observed this in less than 2%.10,11 Based on this lower prevalence, some have advocated that echocardiographic evaluations should not be routinely performed for hEDS in the absence of clinical findings or positive family history. To our knowledge, the present study is the first to report on echocardiographic findings in an older adult population with all hEDS/HSD diagnoses made according to the updated 2017 guidelines. Here, we found aortic dilatation in 15.2% of patients, reaffirming this important association. The prevalence trended higher across increasing age brackets. In the context of previous reports, we conclude that aortic dilatation in hEDS/HSD emerges later in adulthood; echocardiographic assessments may therefore not be necessary in children, but should be included as part of baseline testing for patients ⩾ 21 years. Our data indicate that aortic dimensions in hEDS/HSD remain generally within the mildly dilated range, especially for females, consistent with previous reports. Although interpretation is limited by the retrospective study design, it is unlikely that aortic dilatation in the context of hEDS/HSD poses exaggerated risk of aortic emergency because we included adults over a wide age range and did not find any with aortic dissection/rupture. Of note, two males with hEDS had moderate-to-severe dilatation; aortopathy gene panel sequencing was normal in both cases, with no clear alternative genetic diagnosis. Until the natural history of aortic dilatation can be clarified through large longitudinal studies, patients with hEDS/HSD and aortic dilatation should have ongoing surveillance according to established society guidelines. 21 Of note, we found aortic root dilatation more frequently in patients with hEDS compared to HSDs; because this feature confers a ‘point’ on the hEDS diagnostic checklist, it is challenging to determine whether the risk of aortic pathology is truly higher in hEDS than HSDs.
Extra-aortic arterial involvement (aneurysms, dissections) is frequently cited in the vascular and classical forms of EDS and has not previously been categorically linked to hEDS. We identified systemic arterial complications in five patients with hEDS/HSD: two with CeAD and three with SCAD, one of whom also had a celiac artery pseudoaneurysm. Although the population prevalence of CeAD is unknown, community-based stroke studies have estimated it at one to three per 100,000.22,23 Similarly, the prevalence of SCAD is not known but is estimated at around 0.6–0.24%. 24 Therefore, we report the first description of incidence of CeAD (0.7%) and SCAD (1.8%) in hEDS/HSD, and interpret these phenotypes to be enriched in this cohort. Genetic panel testing was performed based on these clinical indications and did not reclassify the hEDS/HSD diagnoses to an alternative genetic arteriopathy. Vascular medicine specialists should consider hEDS/HSD as part of the differential for these conditions, especially when other underlying conditions such as FMD and genetically defined arteriopathies (i.e., vascular EDS) have been ruled out. Of note, the prevalence of joint hypermobility in FMD is reportedly low, 25 so it should be feasible to clinically differentiate these potential underlying causes. Furthermore, since CeAD may present with neurologic features including headache/migraine or acute neurologic changes, neurologists and headache medicine specialists should be attuned to this association, especially in patients with known hEDS/HSD. 20
The genetic basis of hEDS/HSD is not yet known, but efforts are underway to molecularly characterize these disorders. Through the current clinical classification system and advances in genetic analyses, future investigations may illuminate genetic mediators of aortic and arterial risk in patients with hEDS/HSD. Further study is needed in patients with SCAD and CeAD to determine whether there is a robust association with hEDS/HSD.
Of the 258 individuals included in this study, 245 (95%) were referred to our Cardiovascular Genetics program based on a patient complaint of joint pain or hypermobility or a family history of EDS. Only eight patients were referred based on known arterial abnormalities (aortic dilatation, n = 4; suspected or confirmed SCAD, n = 3; or cervical artery dissection, n = 1). Following the evaluation, 31 individuals received a new diagnosis of thoracic aortic dilatation. Since the vast majority of individuals did not have known cardiovascular abnormalities prior to referral and diagnosis of hEDS/HSD, and because CeAD, SCAD, and peripheral arterial aneurysm/pseudoaneurysm are not part of the diagnostic criteria, we feel confident that this study was not biased towards ascertainment of cardiovascular manifestations in hEDS/HSD. Still, this retrospective study has several limitations inherent to the study design. Although this study contributes important information on the prevalence of aortic dilatation in older adults with hEDS/HSD, there were no longitudinal assessments made and therefore it cannot be used to describe the natural history of this finding. Prospective studies are needed to determine whether the aortic dilatation seen in hEDS/HSD confers an increased risk for future aneurysmal growth and/or dissection. In subjects with extra-aortic findings, diagnoses of SCAD and CeAD were made prior to the time of the hEDS/HSD assessment. Pan-vascular imaging is not routine or recommended in hEDS/HSD, so subclinical incidence of these manifestations could not be robustly ascertained through this study and may not be fully represented. Genetic testing to exclude other genetic connective tissue disorders relied on targeted genes or panels, not genome-wide studies. Lastly, due to lack of objective tests, diagnoses of hEDS/HSD remain clinically challenging.
Conclusion
Cardiovascular manifestations are present in up to 15% of subjects with hEDS and HSD and mainly include mild aortic dilatation and mild MVP. We report, for the first time, that SCAD and CeAD occur, respectively, in 1.8% and 0.7% of patients with hEDS/HSD. Until the natural history of aortic dilatation is fully elucidated, a baseline echocardiographic study in adults under evaluation for hEDS is reasonable and those for whom dilatation is noted should have ongoing surveillance. Patients with extra-thoracic arteriopathy and joint hypermobility should be screened for hEDS/HSD, as these underlying diagnoses are likely under-recognized.
Footnotes
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
This study is supported in part by the National Heart, Lung, and Blood Institute (NHLBI) of the National Institutes of Health (NIH) (ARK: HL140083 and BDG: HL135742). Funders had no role in the design and conduct of the study, in the collection, analysis, and interpretation of the data, or in the preparation, review, or approval of the manuscript.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.