
Abstract
Flow-limiting atherosclerotic lesions of arteries supplying the limbs are a cause of symptoms in patients with peripheral artery disease (PAD). Musculoskeletal metabolic factors also contribute to the pathophysiology of claudication, which is manifest as leg discomfort that impairs walking capacity. Accordingly, we conducted a case–control study to determine whether skeletal muscle metabolic gene expression is altered in PAD. Calf skeletal muscle gene expression of patients with PAD and healthy subjects was analyzed using microarrays. The top-ranking gene differentially expressed between PAD and controls (FDR < 0.001) was PLA2G16, which encodes adipose-specific phospholipase A2 (AdPLA) and is implicated in the maintenance of insulin sensitivity and regulation of lipid metabolism. Differential expression was confirmed by qRT-PCR; PLA2G16 was downregulated by 68% in patients with PAD (p < 0.001). Expression of Pla2g16 was then measured in control (db/+) and diabetic (db/db) mice that underwent unilateral femoral artery ligation. There was significantly reduced expression of Pla2g16 in the ischemic leg of both control and diabetic mice (by 51%), with significantly greater magnitude of reduction in the diabetic mice (by 79%). We conclude that AdPLA is downregulated in humans with PAD and in mice with hindlimb ischemia. Reduced AdPLA may contribute to impaired walking capacity in patients with PAD via its effects on skeletal muscle metabolism. Further studies are needed to fully characterize the role of AdPLA in PAD and to investigate its potential as a therapeutic target for alleviating symptoms of claudication.
Keywords
Introduction
Peripheral artery disease (PAD) affects nearly 10% of people 65 years of age and older, 1 and over 200 million individuals worldwide. 2 Approximately 10–30% of patients with PAD have intermittent claudication, which is defined as leg muscle discomfort that occurs during walking and resolves upon rest. 3 Many others with PAD have impaired walking capacity, characterized by shorter distances walked and slower walking speed. 4 Intermittent claudication results in part from decreased perfusion to the exercising skeletal muscle imposed by flow-limiting atherosclerotic stenosis. However, hemodynamic compromise inadequately explains the degree of functional impairment experienced by patients with PAD, and studies have found a poor correlation between ankle–brachial index (ABI), a measure of perfusion pressure, and walking performance. 5 Moreover, some effective therapies, such as exercise training, improve walking distance in PAD without measurable increases in conduit vessel blood flow.6,7
Therefore, it is likely that non-hemodynamic mechanisms contribute to intermittent claudication and impaired walking capacity in PAD. For example, abnormal skeletal muscle metabolism of nutrients (e.g. glucose and fatty acids) that are critical for adequate adenosine triphosphate and phosphocreatine synthesis could reduce energy availability for exercising muscle. Indeed, we have previously shown that PAD is associated with insulin resistance and reduced uptake of glucose into skeletal muscle.8,9 Notably, insulin resistance is linked to impaired mitochondrial function in skeletal muscle10,11 and to downregulation of critical genes that regulate fatty acid oxidation, glucose metabolism, and oxygen consumption.12–15 Therefore, in the present study, we sought to identify alterations in skeletal muscle gene expression that could contribute to the pathophysiology of intermittent claudication and impaired walking capacity in patients with PAD.
Materials and methods
Patient population
A diagnosis of PAD was based on symptoms of intermittent claudication and a resting ABI of ⩽ 0.90 and/or a decrease in ABI of at least 20% following treadmill exercise. PAD subjects were excluded if they had a physician diagnosis of diabetes mellitus or plasma glucose > 200 mg/dL during a 2-hour oral glucose tolerance test (OGTT); signs of critical limb ischemia (rest pain, ulceration, gangrene); peripheral vascular surgery, percutaneous coronary intervention, myocardial infarction, carotid artery surgery, stroke or transient ischemia attack within the last 6 months; changes in dosage of pentoxifylline, cilostazol or HMG-CoA reductase inhibitors within 3 months; or exercise limitations other than claudication (caused by heart failure, angina, chronic obstructive pulmonary disease (COPD), arthritis, neuropathy, etc.). Forty patients with PAD and stable claudication symptoms consented to participate. Of these, five did not qualify after further evaluation for inclusion and exclusion criteria (two with abnormal OGTT, two were unable to stop aspirin or clopidogrel prior to muscle biopsy, one with post-exercise ABIs > 0.90), six voluntarily withdrew, and one could not continue participation due to hospitalization unrelated to the study. Healthy subjects were at least 50 years of age, had no known medical problems, and were ineligible if they had smoked within the past year. Nineteen healthy control subjects consented to participate. Of these 19 healthy subjects, five did not qualify due to abnormal lipid profiles, and two were lost to follow-up prior to muscle biopsy. Subjects were recruited from clinical practices at Brigham and Women’s Hospital and by advertisement. This study was approved by the Institutional Review Board at Brigham and Women’s Hospital, and all subjects gave written informed consent.
Calf muscle biopsy
Biopsies from the medial head of the gastrocnemius muscle were obtained from the patients with PAD and the healthy control subjects. After local anesthesia with lidocaine, approximately 100–150 mg of muscle tissue was collected, using a 5 mm Bergstrom percutaneous muscle biopsy needle. For PAD subjects, samples were taken from the more symptomatic leg or the leg with a lower ABI if the subject experienced equal symptoms bilaterally. Samples were taken from either leg of the healthy subjects. Samples were immediately frozen in liquid nitrogen and stored at –80°C until further analysis.
Gene expression quantification and analysis
A portion of each muscle biopsy was homogenized (IKA, Staufen, Germany), and total RNA was isolated using TRIzol® (Invitrogen™, Carlsbad, California). RNA quality was assessed using Bioanalyzer (Santa Clara, California). Samples from four participants (two PAD, two healthy) were excluded due to poor RNA quality owing to degradation. cDNA was synthesized and transcribed in vitro using biotinylated nucleotides to generate labeled cRNA, using the Affymetrix 3’ IVT PLUS Reagent Kit (Waltham, Massachusetts). Following purification and fragmentation, cRNA samples were hybridized to Affymetrix GeneChip PrimeView Human Gene Expression Arrays; microarrays were washed and scanned using standard protocols. Quality control analysis of microarray data revealed that one PAD sample was an outlier. With exclusion of these samples, a total of 25 PAD and 10 healthy samples were utilized for analysis. Microarray data were normalized using robust multi-array average, and data were log2 scale-transformed. Probesets were analyzed for differential expression using linear modeling in the R/Bioconductor package limma,16–18 and false discovery rates (FDR) were calculated to account for multiple testing. Data are available at Gene Expression Omnibus (GEO GSE113873).
Expression of PLA2G16 between PAD and healthy participants was analyzed using quantitative real-time polymerase chain reaction (qRT-PCR) on cDNA synthesized with the High Capacity cDNA Reverse Transcription kit (Applied Biosystems, Waltham, MA, USA) with SybrGreen detection [ABI 7900; Applied Biosystems (Thermo Fisher Scientific, Waltham, Massachusetts)] (primer sequences provided in online Supplementary Table 1, available at https://doi.org/10.6084/m9.figshare.11974479).
Whole-body and calf skeletal muscle insulin sensitivity
A hyperinsulinemic-euglycemic clamp was performed in 10 patients with PAD and three healthy control subjects to evaluate whole-body insulin sensitivity and skeletal muscle glucose uptake, as previously described. 8 Briefly, under fasting conditions, subjects were given a primed weight-based insulin infusion (2 mU/kg/min). Plasma glucose was measured at 5-minute intervals. Dextrose infusion (20%) was adjusted to maintain glucose of approximately 4.4 mmol/L (80 mg/dL). Steady state was achieved when the dextrose infusion rate varied by no more than 5% between consecutive measurements. Whole-body insulin sensitivity was assessed as the glucose disposal rate (M) from the glucose infusion rates over the last 20 minutes of the clamp. Skeletal muscle glucose uptake was evaluated using fluorodeoxyglucose (FDG-PET) imaging as previously described. 8 Imaging was performed using a whole-body PET scanner (Discovery LS PET/CT Imaging System; GE, Milwaukee, WI, USA) that acquires 47 contiguous transaxial planes with an image resolution of 5.4 mm at full-width–half-maximum in-plane and 5.4 mm at full-width–half-maximum in the axial direction. The overall rate of calf muscle FDG uptake (Ki) was quantified using the graphical Patlak analysis. Net glucose uptake was determined from the overall rate of FDG uptake multiplied by the average arterial glucose level during the scan. The resulting rate of glucose uptake for each muscle group is expressed in µmol/min per kilogram of tissue.
Mouse studies
In order to investigate potential mechanisms contributing to altered expression of PLA2G16 in humans with PAD and intermittent claudication, exploratory studies were performed to analyze the expression of Pla2g16 in mouse models of dietary and pharmacologic insulin resistance, and in diabetic db/db and control db/+ mice who underwent unilateral femoral artery ligation. All animal studies were approved by respective Institutional Animal Care and Use Committees at the Joslin Diabetes Center and Brigham and Women’s Hospital. Mice were purchased from Jackson Laboratories (Bar Harbor, ME, USA).
For dietary manipulation studies, female C57BL/6J mice were randomly assigned to either a 60% fat diet (60% of calories from fat, n = 5) to induce obesity and insulin resistance, or a 10% fat diet (control, n = 5) (Research Diets D1249 or D12450, respectively) for 48 weeks. For analysis of pharmacologically induced insulin resistance, the peptide insulin receptor antagonist S961 (Phoenix Pharmaceuticals, Mannheim, Baden-Württemberg, Germany) was administered via an Alzet® osmotic minipump (Durect, Cupertino, California), implanted subcutaneously in the dorsal interscapular region after anesthesia with tribromoethanol, for 7 days to induce acute insulin resistance in male C57BL/6J mice, as described. 19 Five mice received 5 nmoles of S961, while five received 10 nmoles of S961, and five mice were infused with phosphate-buffered saline (PBS) as control. Sample sizes were estimated based on results of prior studies19,20 and by confirmation of induction of insulin resistance in the treated cohorts using insulin tolerance testing after a 4-hour fast (regular human insulin, 0.75 U/kg intraperitoneally). 21 Mice were sacrificed after full anesthesia with pentobarbital (50 mg/kg, intraperitoneal).
Ischemic injury was produced by unilateral femoral artery ligation in 10-week-old male diabetic db/db and normoglycemic control db/+ mice, as previously described, and the sample size was estimated based on results of a prior study.22,23 Mice were imaged 3 (n = 11,13), 11 (n = 5,6), and 31 (n = 4,7) days following surgery with a 785-nm near-infrared Laser Doppler Imager-2 (LDI2; Moor Instruments Inc., Wilmington, DE). The mean blood flow percentage was determined by comparing the injured to uninjured leg (LDI V5.3). These mice were sacrificed after euthanasia with CO2, and skeletal muscles were harvested to measure expression of Pla2g16 in the ischemic and non-ischemic gastrocnemius skeletal muscles at the indicated time points after femoral artery ligation.
Differential expression of Pla2g16 expression in mouse muscle was analyzed by quantitative PCR, as described above for human studies. Primer sequences are provided in online Supplementary Table 1.
Statistical analysis
Descriptive and experimental characteristics are reported as the mean and SD for continuous variables and percent for categorical variables. Differences between patients with PAD and the healthy subject groups were compared using unpaired t-tests. Differences for categorical variables were compared using chi-squared tests. Multivariate linear regression was utilized to evaluate for independent predictors of adipose-specific phospholipase A2 (AdPLA) expression; those baseline variables that were significantly different between the PAD and healthy subject groups were entered into the regression model (Table 1). For exploratory mouse studies, between-group differences were analyzed using unpaired t-tests. For human gene expression analyses, FDR < 0.01 and p < 0.05 (two-tailed) were considered significant. For all other data comparisons, p < 0.05 were considered significant. Statistical analyses were performed using SPSS v.23 (IBM, Armonk, NY, USA) and GraphPad Prism v.7 (GraphPad Software, Inc., La Jolla, CA, USA).
Baseline subject characteristics.
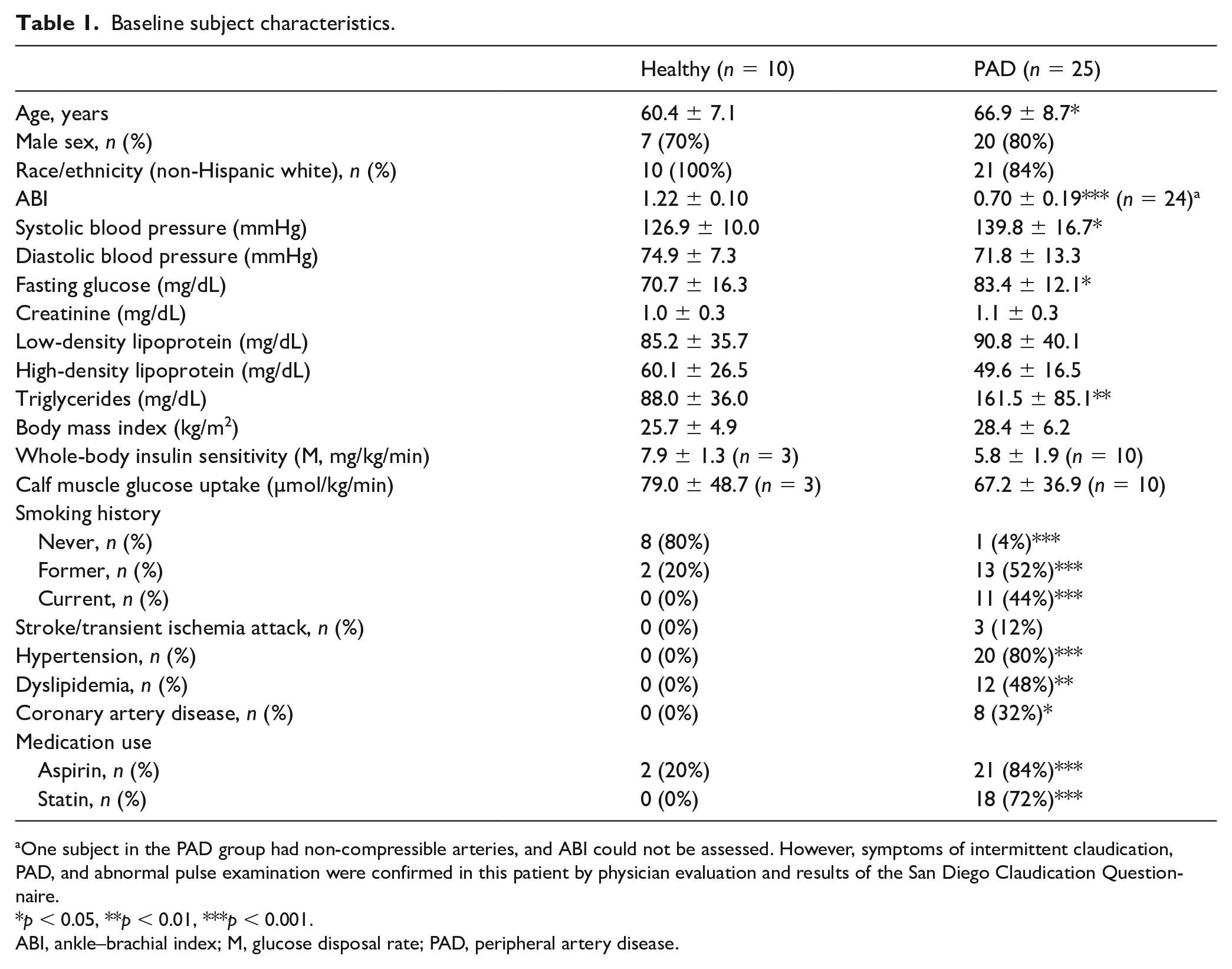
One subject in the PAD group had non-compressible arteries, and ABI could not be assessed. However, symptoms of intermittent claudication, PAD, and abnormal pulse examination were confirmed in this patient by physician evaluation and results of the San Diego Claudication Questionnaire.
p < 0.05, **p < 0.01, ***p < 0.001.
ABI, ankle–brachial index; M, glucose disposal rate; PAD, peripheral artery disease.
Results
Baseline characteristics of study participants
Baseline characteristics of the 25 subjects with PAD and 10 healthy control subjects who were included in the microarray analysis are presented in Table 1. The mean ABI in PAD subjects was 0.70 ± 0.19. Compared to healthy subjects, PAD subjects were slightly older, had higher systolic blood pressure, fasting glucose, and plasma triglycerides. PAD subjects also had higher prevalence of past and current smoking, as well as hypertension, and coronary artery disease. The majority of subjects with PAD were taking aspirin and statins. Insulin sensitivity, as measured by hyperinsulinemic-euglycemic clamp, was lower, albeit not significantly, in patients with PAD compared to healthy subjects; similarly, calf muscle glucose (FDG) uptake tended to be lower.
Impact of PAD on skeletal muscle gene expression
Quality control metrics revealed that the gene expression profiles for muscle samples obtained from both groups of participants displayed the expected distribution of expression across probe sets included in the microarray. Principal component analysis (PCA) revealed lack of separation in PAD versus control groups, indicating substantial variability between participants independent of group assignment (PCA plot; Supplementary Figure 1 available at https://doi.org/10.6084/m9.figshare.11974398).
Differential expression between groups was assessed, and the level of significance was plotted versus fold change between groups (volcano plot, Figure 1). The top-ranking gene was PLA2G16, the gene encoding AdPLA, which demonstrated a 69% reduction in expression in PAD as compared with controls (FDR < 0.001) (Figure 2A). The diagnosis of PAD was the only variable associated with lower AdPLA expression (p = 0.026) in the multivariate linear regression model. In addition, qPCR analysis in a subset of nine controls and 15 patients with PAD (for whom sufficient RNA was available) confirmed reduced expression of PLA2G16 in PAD (68% lower, p < 0.001) (Figure 2B).
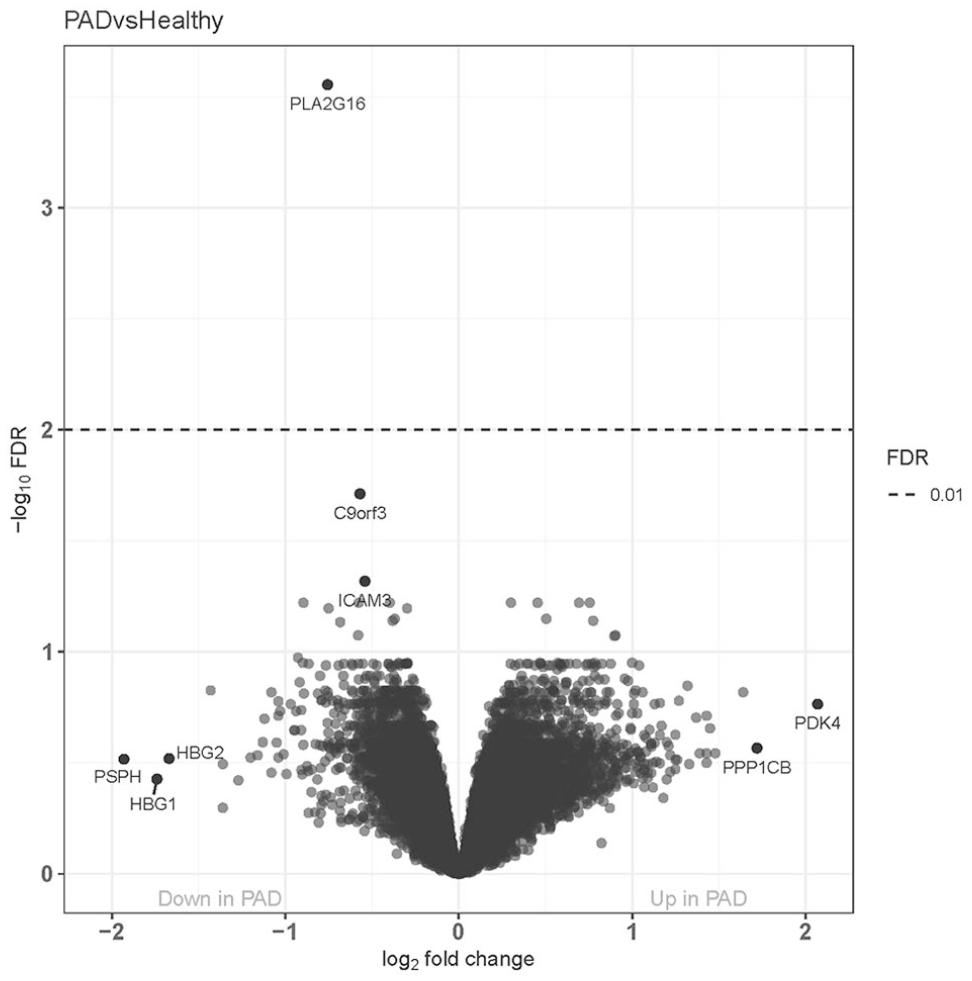
Volcano plot of differential gene expression. Of the 49,396 total probe sets analyzed, only one probe set met our threshold for significance for differential expression between subjects with patients with PAD and healthy subjects.
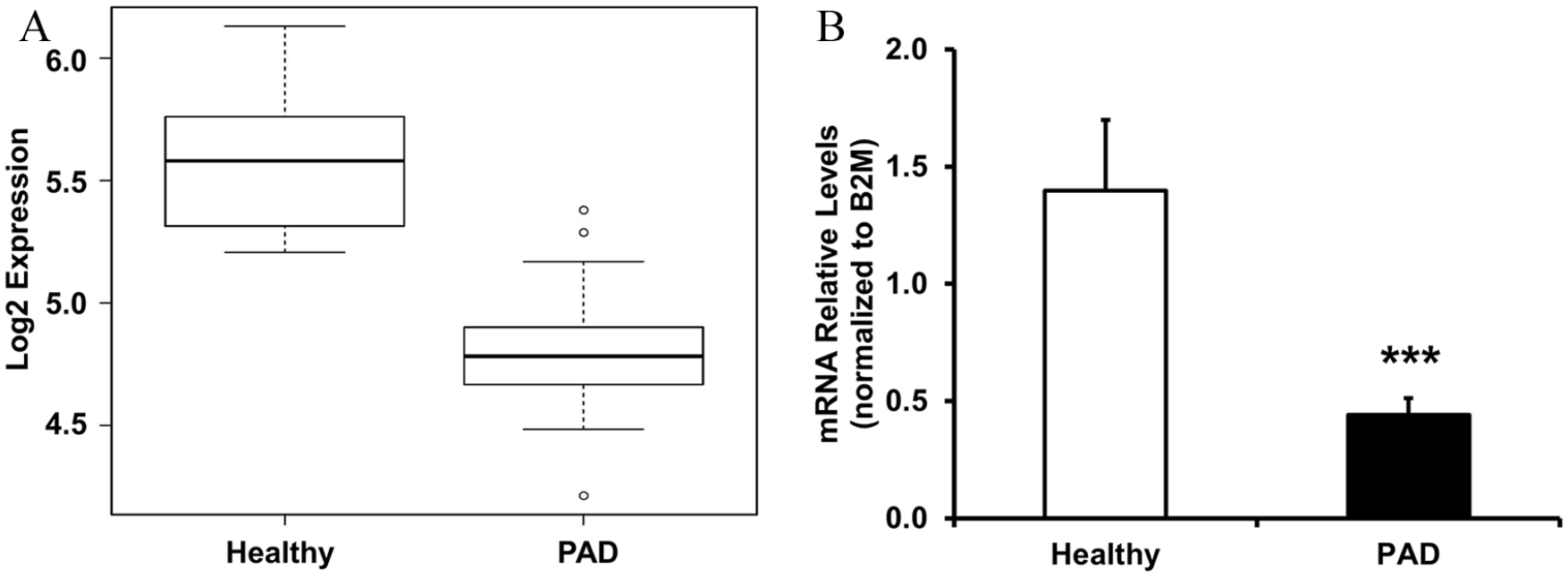
PLA2G16 gene expression. (A) Expression for PLA2G16 (probe set 11741020_a_at) in healthy subjects and patients with PAD; FDR < 0.01. (B) qPCR analysis of PLA2G16 expression in gastrocnemius muscle biopsies from healthy subjects (n = 9) and patients with PAD (n = 15), normalized to B2M; ***p < 0.001.
Two additional transcripts, C9orf3 (also known as aminopeptidase 0) and ICAM3, were also downregulated in PAD (48% and 45%, respectively, FDR < 0.05). Expression patterns for the top 50 differentially expressed probesets are presented in the heat map in Figure 3 and online Supplementary Table 2. Among the transcripts of interest were: (1) multiple probesets for the serine/threonine phosphatase regulatory subunit PPP2R1A, upregulated by 1.5–2-fold in PAD (nominal p < 0.0001, FDR < 0.12); (2) multiple probe sets for TXNIP (thioredoxin interacting protein), upregulated by 1.5–1.7-fold (nominal p < 0.0001, FDR < 0.12); and (3) PDK4 (pyruvate dehydrogenase kinase 4), which phosphorylates and inhibits pyruvate dehydrogenase, upregulated by more than fourfold in PAD (nominal p < 0.001, FDR 0.17).
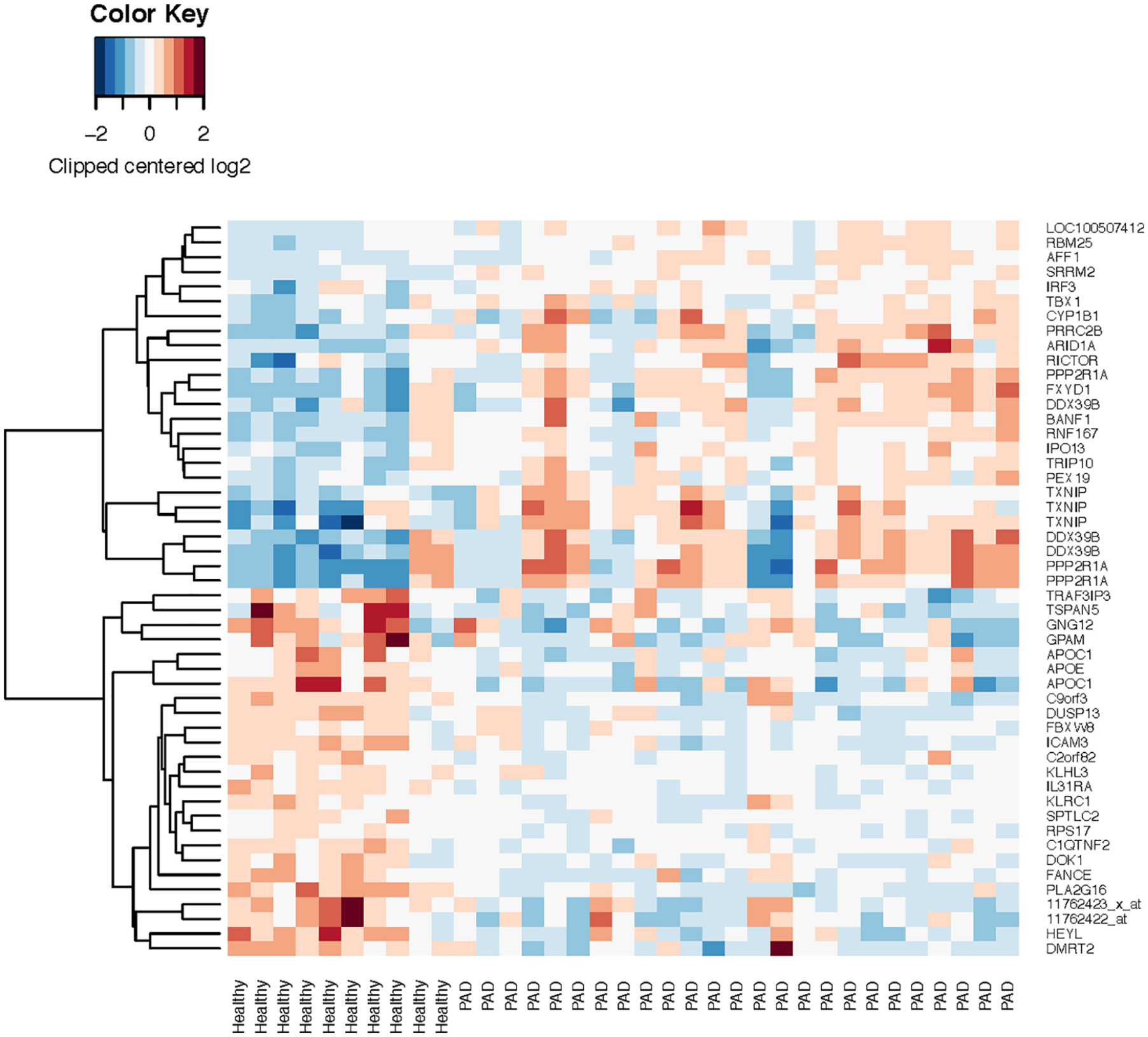
Heat map of expression patterns.
Impact of insulin resistance and ischemic injury on expression of muscle Pla2g16
The reduction in PLA2G16 in individuals with PAD relative to healthy controls could be linked to altered systemic metabolism and/or the tissue insulin resistance characteristic of PAD. Accordingly, we evaluated the effect of dietary and pharmacologic insulin resistance on expression of Pla2g16 in murine hindlimb muscle. Expression of Pla2g16 did not differ in gastrocnemius muscle isolated from mice fed a high-fat versus low-fat diet (60% vs 10% calories from fat, Figure 4A). Likewise, infusion of S961, an insulin receptor inhibitory peptide, did not alter expression of Pla2g16 in quadriceps muscle (Figure 4B), despite induction of systemic insulin resistance and hyperinsulinemia (not shown). Moreover, there was no difference in the expression of Pla2g16 in the uninjured leg of diabetic db/db mice, as compared with control db/+ mice (Figure 5A).
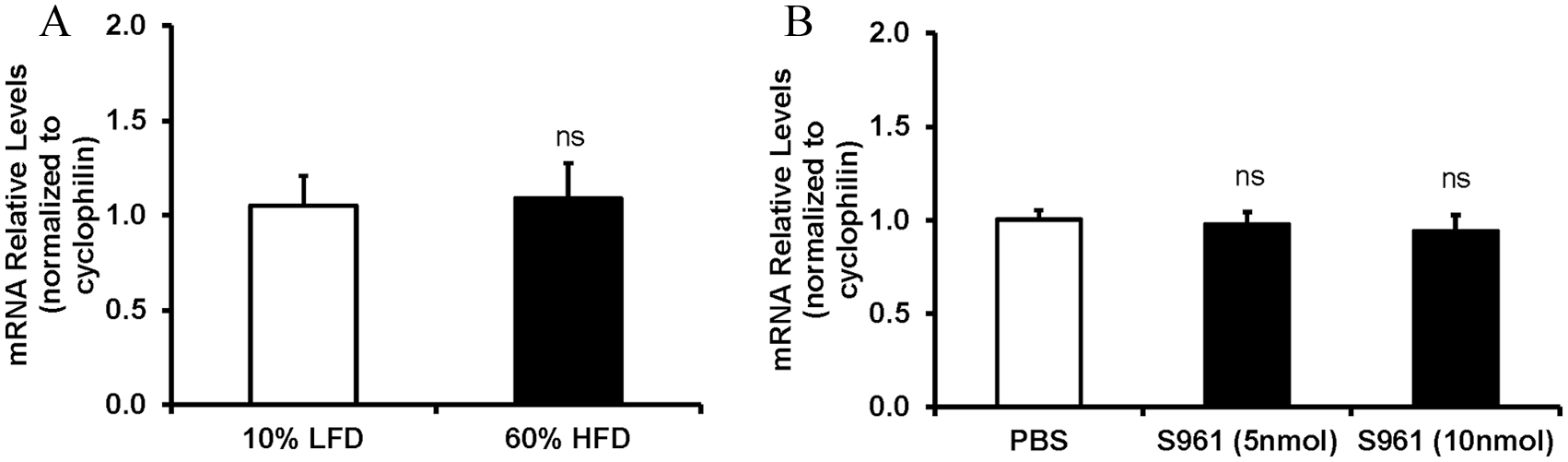
Effects of dietary and pharmacologic insulin resistance on expression of Pla2g16 in murine hind limb muscle: (A) qPCR analysis of Pla2g16 expression in gastrocnemius muscle of mice treated with LFD (10%, n = 5) and HFD (60%, n = 5), normalized to cyclophilin. (B) qPCR analysis of Pla2g16 expression in quadriceps muscle of mice treated with PBS, or the insulin receptor antagonist S961, at 5 or 10 nmol dose, normalized to cyclophilin.
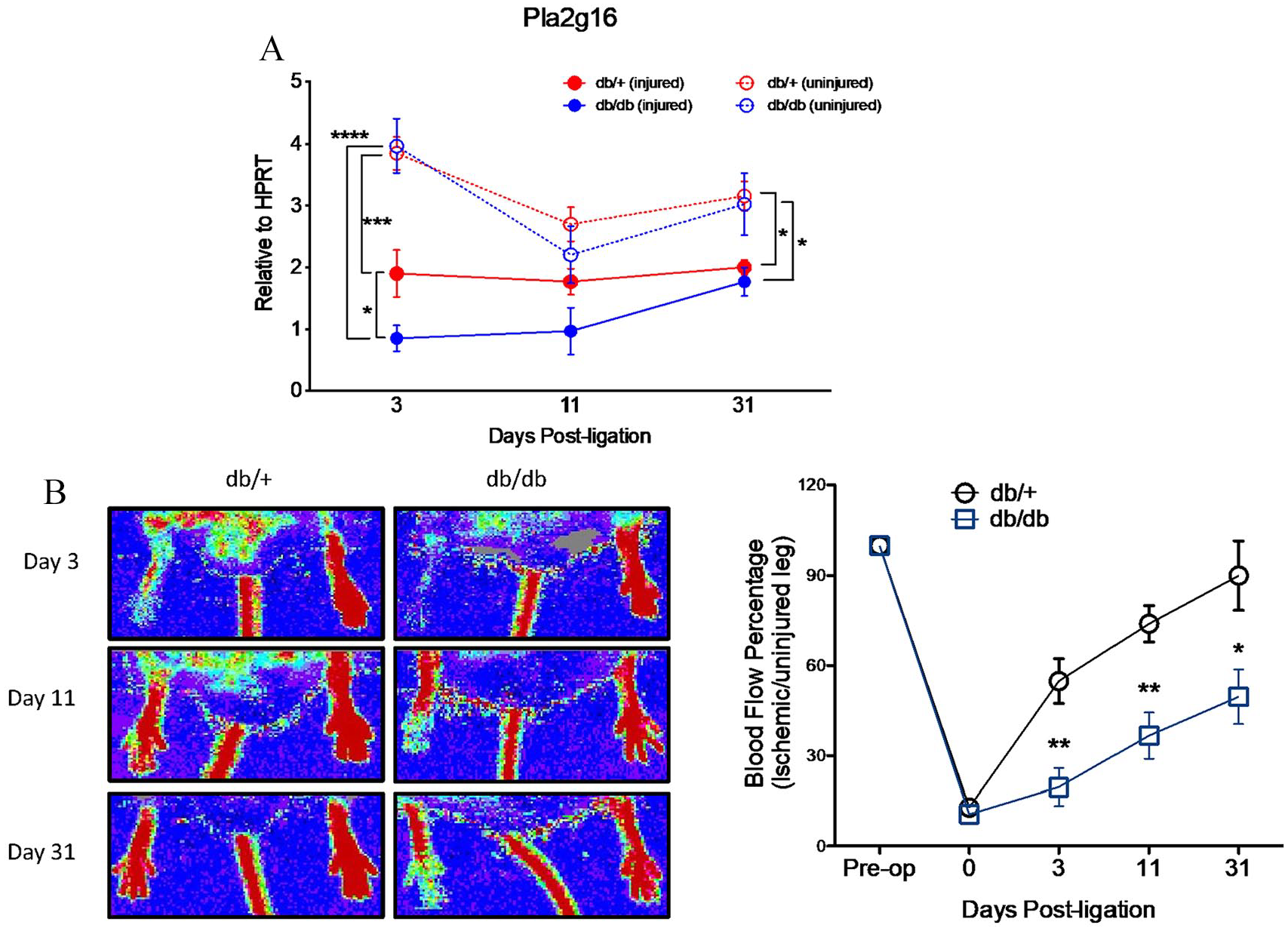
Effect of hindlimb ischemia in Pla2g16 expression and blood flow in diabetic and nondiabetic mice: (A) qPCR analysis of Pla2g16 expression in gastrocnemius muscle of the injured and uninjured leg of db/+ and db/db mice subjected to femoral artery ligation after 3 (n = 11, 13), 11 (n = 5, 6), and 31 days (n = 4, 7), normalized to Hprt. (B) Left: representative laser Doppler images displaying recovery of blood flow in ligated right hindlimb between db/+ and db/db mice. Right: quantification of mean blood flow at days 3, 11, and 31 post-injury.
To test the impact of ischemia on expression of Pla2g16, we performed femoral artery ligation, which acutely reduced blood flow in the affected leg in both db/+ and db/db mice (Figure 5B). Blood flow recovery after femoral artery ligation was significantly impaired in db/db mice compared to control db/+ mice. In both db/+ and db/db mice, there was significantly reduced expression of Pla2g16 in the gastrocnemius muscle of the leg subject to acute ischemic injury by femoral artery ligation as compared to the uninjured leg (by 51%). This decrease in expression was significant as early as 3 days after ligation, and persisted at 11 and 31 days after ligation (Figure 5A). The reduction in expression of Pla2g16 in the leg subject to femoral artery ligation was significantly greater in db/db mice compared with db/+ mice, most notably at day 3 (by 79%), but the difference dissipated by day 31.
Discussion
We report a novel observation that patients with PAD have significant downregulation of PLA2G16, the gene encoding AdPLA, in lower limb skeletal muscle. Pla2g16 also was reduced in mice subjected to acute ischemic injury following ligation of the femoral artery, with a greater downregulation in diabetic mice compared to non-diabetic mice. By contrast, expression of Pla2g16 was not altered in mice in response to diet-induced insulin resistance or pharmacologic inhibition of the insulin receptor, or in the normal perfused leg of db/db mice compared with control mice. Collectively, these findings suggest that reduced expression of PLA2G16 in human PAD is likely a consequence of disordered transcriptional regulation in the setting of decreased blood flow or other features of the PAD environment.
AdPLA expression and action
AdPLA protein was initially identified as HRASLS3 (Ha-RAS-like suppressor 3 or H-Rev-107-1), a class II tumor suppressor. 24 Although it was initially reported to be ubiquitously expressed at low levels,25,26 it was later named and characterized based on its high expression in mouse adipose tissue.27,28 Its expression also has been detected in multiple human tissues, including skeletal muscle.26,27,29 Phospholipases are a diverse group of proteins, all of which can hydrolyze membrane phospholipids. Phospholipase A2s (PLA2s) specifically release fatty acids from the second carbon of the glycerol backbone, and since arachidonic acid is most common at this position, PLA2s catalyze a rate-limiting step in arachidonic acid production. 30 AdPLA is the only known member of phospholipase A2 group XVI, a recently discovered group of calcium-dependent intracellular PLA2s. 27 AdPLA is also unique among PLA2 family members in its catalytic mechanism, utilizing a cysteine-histidine-histidine motif at its active site. 31 AdPLA is distinct from lipoprotein-associated PLA2, for which increased activity is associated with elevated risk for stroke and coronary artery disease.32–34
While we do not know the specific mechanisms responsible for downregulation of PLA2G16 in patients with PAD, previous studies have demonstrated dysregulation of PLA2G16 in response to various physiologic conditions relevant to PAD. For example, PLA2G16 is decreased by 71% in human monocyte-derived dendritic cells exposed to hypoxic conditions (GEO GSE22282). Similarly, expression in mouse myotubes is downregulated by 55% after 48 hours of serum starvation (GEO GSE1776) 35 and by 56% in response to treatment with the saturated fatty acid palmitate (GEO GSE6766). 36 Conversely, homocysteine treatment increases expression of Pla2g16 in mouse gonadal adipose tissue and 3T3-L1 adipocytes; changes in expression are accompanied by increased PLA2 activity and generation of lysophosphatidylcholine species linked to activation of the inflammasome. 37 Interestingly, expression patterns are not altered by age (GEO GSE362), fasting (GEO GSE46495), type 2 diabetes (in either myotubes or arterial tissue, GEO GSE12643, GEO GSE13760), weight loss (GEO GSE17371), or obesity (GEO GSE48964). Additional regulation of PLA2G16 may be mediated by miRNA. Yang et al. identified Pla2g16 as a potential target of miR-1996, which was increased nearly twofold in murine skeletal muscle in response to short-term ischemia/reperfusion. 38 Thus, regulation of PLA2G16 is clearly complex, and is likely to be tissue-specific, time-dependent, and context-dependent. Nevertheless, our data indicate that direct experimental induction of ischemia in vivo results in inhibition of Pla2g16. Understanding the mechanisms mediating transcriptional regulation of PLA2G16 and the impact of downstream lipid species altered in response to PLA2G16-mediated activity will be an important goal for future experiments.
Pathophysiologic mechanisms linking AdPLA to walking impairment
AdPLA2 downregulation, as observed in our patients with PAD, could play a causal role in the insulin resistance that we have characterized previously as a feature of PAD.8,9 Indeed, AdPLA-null mice develop whole-body insulin resistance and dysregulation of lipolysis within adipose tissue. 28 Loss of insulin sensitivity resulting from a decrease in AdPLA expression likely stems from a decrease in the availability of arachidonic acid for synthesis of downstream products. 39 Though the impact of this gene in skeletal muscle is uncertain, arachidonic acid and its eicosanoid byproducts have been implicated in several key processes associated with metabolism, including insulin regulation of glucose uptake. For example, treatment with arachidonic acid increases translocation of GLUT1 and GLUT4 to the plasma membrane, boosting adipocyte glucose uptake. 40 Additionally, treatment of isolated rat skeletal muscle cells with arachidonic acid ameliorates insulin resistance and increases glucose uptake. 41 Thus, it is possible that decreased expression of PLA2G16 in patients with PAD would lead to reduced production of arachidonic acid and downstream eicosanoid mediators, and thereby contribute to insulin resistance in PAD. Alternatively, insulin resistance in the patients with PAD might have caused decreased expression of PLA2G16. Our mechanistic studies in mice support both possibilities. In the mouse models of insulin resistance in which there was no disturbance of limb blood flow, there was no alteration of Pla2g16 expression, suggesting that insulin resistance was likely the result, and not the cause, of decreased PLA2G16 expression levels in patients with PAD. However, the ischemic skeletal muscles of our db/db mice exhibited the lowest expression of Pla2g16, suggesting that insulin resistance, hyperglycemia, increased adiposity, or other features of the dysregulated metabolism characteristic of leptin-resistant db/db mice might further exacerbate the reduction in PLA2G16 expression in the setting of ischemia associated with PAD.
AdPLA2 downregulation could also result in a state of chronic inflammation, exacerbating symptoms, reducing activity, and further disrupting skeletal muscle metabolism. We and others have found that biomarkers of vascular inflammation are associated with functional measures in patients with PAD.42,43 Resolution of inflammation is largely regulated by lipid mediators, including lipoxin A4 (LXA4), which is synthesized from metabolites of arachidonic acid via the 12/15-LO pathways.44–46 LXA4 promotes resolution by reducing recruitment of neutrophils to the site of inflammation and by stimulating phagocytosis of apoptotic leukocytes by macrophages. 45 Plasma levels of aspirin-triggered lipoxin, an epimer of LXA4 synthesized by aspirin-acetylated cyclooxygenase-2, are inversely correlated with the severity of PAD. 47 When compared to age-matched controls, patients with intermittent claudication have a 40% reduction in circulating aspirin-triggered lipoxin. The downregulation of PLA2G16 that we detected, and subsequent downregulation of arachidonic acid, could provide an explanation for the observed reduction in lipoxin production observed previously in patients with PAD. This diminished availability of lipoxins would facilitate a chronic state of inflammation.
We recognize that additional genes and regulatory networks are likely to contribute to the molecular pathophysiology of PAD at a tissue level. Gene ontology analysis of the top 20 differentially expressed genes in PAD (FDR < 0.1) in our study identified neutral lipid metabolic processes as the top ranking significant pathway, with downregulation of PLA2G16, APOC1, and APOE responsible for enrichment (online Supplementary Table 2). Additionally, GPAM, the first step in glycerolipid synthesis, is also significantly downregulated. Together, these data highlight perturbations in lipid metabolism as a major phenotype skeletal muscle in patients with PAD. These findings are of particular interest given recent findings from a genome-wide, multi-ancestry type 2 diabetes-association meta-analysis of 228,499 cases and 1,178,783 controls from the Million Veteran Program, DIAMANTE, Biobank Japan, and other studies, 48 which identified a SNP near PTDSS1 (rs3104154) as linked to PAD. Like PLA2G16, PTDSS1 is also involved in glycerolipid metabolism (KEGG pathway) and is responsible for catalyzing the formation of phosphatidylserine from phosphatidylcholine or phosphatidylethanolamine. Whether the genetic variation at PTDSS1 contributes to risk for PAD or the established PAD transcriptomic phenotype will require additional study. In another recent study, Ryan et al. performed whole transcriptome sequencing and mitochondrial phenotyping of calf muscle from patients with intermittent claudication, critical limb ischemia, and non-PAD controls and reported that critical limb ischemia, but not intermittent claudication, was associated with altered expression of nuclear-encoded OXPHOS mitochondrial genes, with parallel reductions in mitochondrial respiratory capacity. RNA-seq analysis reported in the data set (GEO GSE120642) of this study found that expression of PLA2G16 was modestly increased in the patients with intermittent claudication, and decreased in the patients with critical limb ischemia. 49
Limitations
There were several minor, but expected, differences between patients with PAD and the healthy control subjects. Patients with PAD were slightly older, had higher systolic blood pressure, glucose and triglyceride levels, a greater prevalence of smoking, and other vascular disease, and most were taking aspirin and a statin medication, as typifies this population. However, after controlling for characteristics that differed significantly between the two groups, the diagnosis of PAD remained an independent predictor of lower AdPLA expression. Also, we did not determine the specific cell composition for each calf muscle biopsy. We acknowledge that skeletal muscle tissue is heterogeneous, and biopsy samples can include mature myotubes, satellite cells, and intramuscular fat. PLA2G16 has been shown to be widely expressed, including in muscle cells and in adipocytes. Future experiments could utilize single cell analysis to define the precise subpopulations of cells responsible for altered transcriptomic patterns in PAD.
Conclusions
In summary, we have identified significant downregulation of PLA2G16 in patients with PAD compared to healthy controls. We observed similarly a downregulation of Pla2g16 in mice with hindlimb ischemia, which was most profound in diabetic mice. Thus, reduced expression of PLA2G16 in human PAD is likely a consequence of disordered transcriptional regulation in the setting of decreased blood flow. Decreased AdPLA may contribute to impaired walking capacity in patients with PAD via its effects on insulin sensitivity or other aspects of skeletal muscle metabolism. Further studies are needed to fully characterize the role of AdPLA in PAD and to investigate its potential as a therapeutic target for alleviating symptoms of claudication.
Supplemental Material
Supplementary_Figure_1 – Supplemental material for Skeletal muscle expression of adipose-specific phospholipase in peripheral artery disease
Supplemental material, Supplementary_Figure_1 for Skeletal muscle expression of adipose-specific phospholipase in peripheral artery disease by Caitlin Parmer, Ana Luisa De Sousa-Coelho, Henry S Cheng, Grace Daher, Alison Burkart, Jonathan M Dreyfuss, Hui Pan, Joshua C Prenner, Jessica M Keilson, Reena Pande, Stanislav Henkin, Mark W Feinberg, Mary Elizabeth Patti and Mark A Creager in Vascular Medicine
Footnotes
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed the following financial support for the research, authorship, and/or publication of this article: Dr De Sousa-Coelho was supported by a mentor-based postdoctoral fellowship grant from the American Diabetes Association (to MEP). Dr Pande was supported by a Research Career Development Award (K12HL083786) from the National Heart, Lung, and Blood Institute, and a Scientist Development Grant (10SDG4200060) from the American Heart Association. Drs Patti, Dreyfuss, and Pan acknowledge support from the Joslin Diabetes Research Center (P30 DK036836). Drs Cheng, Feinberg, and Creager were supported by a Strategically Focused Research Center Award (18SFRN33900085) from the American Heart Association.
Supplementary material
The supplementary material is available online with the article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.