
Abstract
Vascular malformations occur during early vascular development resulting in abnormally formed vessels that can manifest as arterial, venous, capillary or lymphatic lesions, or in combination, and include local tissue overdevelopment. Vascular malformations are largely caused by sporadic somatic gene mutations. This article aims to review and discuss current molecular signaling pathways and therapeutic targets for vascular malformations and to classify vascular malformations according to the molecular pathways involved. A literature review was performed using Embase and Medline. Different MeSH terms were combined for the search strategy, with the aim of encompassing all studies describing the classification, pathogenesis, and treatment of vascular malformations. Major pathways involved in the pathogenesis of vascular malformations are vascular endothelial growth factor (VEGF), Ras/Raf/MEK/ERK, angiopoietin-TIE2, transforming growth factor beta (TGF-β), and PI3K/AKT/mTOR. These pathways are involved in controlling cellular growth, apoptosis, differentiation, and proliferation, and play a central role in endothelial cell signaling and angiogenesis. Many vascular malformations share similar aberrant molecular signaling pathways with cancers and inflammatory disorders. Therefore, selective anticancer agents and immunosuppressants may be beneficial in treating vascular malformations of specific mutations. The current classification systems of vascular malformations, including the International Society of the Study of Vascular Anomalies (ISSVA) classification, are primarily observational and clinical, and are not based on the molecular pathways involved in the pathogenesis of the condition. Several molecular pathways with potential therapeutic targets have been demonstrated to contribute to the development of various vascular anomalies. Classifying vascular malformations based on their molecular pathogenesis may improve treatment by determining the underlying nature of the condition and their potential therapeutic target.
Introduction
Vascular malformations have a worldwide prevalence of 0.3–1.5%. 1 Vascular malformations occur during early vascular development, resulting in abnormally formed vessels that can manifest as arterial, venous, capillary or lymphatic lesions, or in combination, and include local tissue overdevelopment. These lesions are largely caused by sporadic somatic gene mutations 2 that regulate vital pathways such as angiogenesis, apoptosis, maturation, proliferation, and growth of vascular cells. 3 Vascular malformations are presumed to be present at birth but remain subclinical until presentation. They tend to be static or slow in growth in proportion to the child, and do not involute spontaneously. However, certain physiological and pathological circumstances, such as hormonal change (e.g. puberty or pregnancy) and trauma, respectively, are recognized to accelerate their growth and proliferation. Patients may complain of the disfiguring appearance and/or functional impairment caused by the vascular malformation that could lead to psychological distress. Complications can be localized to the lesion, such as pain, bleeding and infection, and be systemic, such as pulmonary embolism, limb loss and life-threatening end-organ failure including high-output cardiac failure. 4 The patients have also been shown to have poorer quality of life than the general population. 5
In 2014 and 2018, the International Society of the Study of Vascular Anomalies (ISSVA) updated the existing classification with new information on the genetics and histology of vascular anomalies, dividing the tumor group into benign, locally aggressive or borderline, and malignant. Vascular malformations were divided into four groups: simple, combined, those of major named vessels (lymphatics, veins, arteries), and those associated with other anomalies (Table 1). 6
Overview of classification of vascular anomalies according to 2018 ISSVA guidelines6.

Adapted from Ref 6 with permission from ISSVA as per the Creative Commons Attribution 4.0 Interatnional License (https://creativecommons.org/licenses/by/4.0/).
CLM, capillary lymphatic malformation; ISSVA, International Society of the Study of Vascular Anomalies; KTS, Klippel–Trenaunay syndrome.
The current classification systems, especially the ISSVA classification, have been widely used by both clinicians and scientists. However, despite the ISSVA classification listing associated causal genes, the classification system is primarily observational and clinical, and not based on the molecular pathways involved in the pathogenesis of the condition. This article aims to review and discuss current understanding of the molecular signaling pathways and potential therapeutic targets for vascular malformations and to classify vascular malformations according to the molecular pathways involved.
Methods
A literature search was performed on Embase (1980–2019) and Medline (1980–2019). The MeSH terms used in Embase were ‘congenital blood vessel malformation’, ‘classification’, ‘genetics’, and ‘drug therapy’. The MeSH terms used in Medline were ‘vascular malformations’, ‘classification’, ‘genetics’, and ‘drug therapy’. Only full articles in English that reported on vascular malformations were included. Unpublished material, abstracts, and letters were all excluded.
Results
Mechanisms of pathogenesis
Vascular morphogenesis is divided into vasculogenesis and angiogenesis (Figure 1). Vasculogenesis begins in the extra-embryonic mesoderm of the yolk sac 7 and involves three main phases. The first phase is initiated from the generation of hemangioblasts; the second phase involves the proliferation and differentiation of angioblasts into endothelial cells; and the third phase is the formation of primary capillary plexus from endothelial cells.8,9
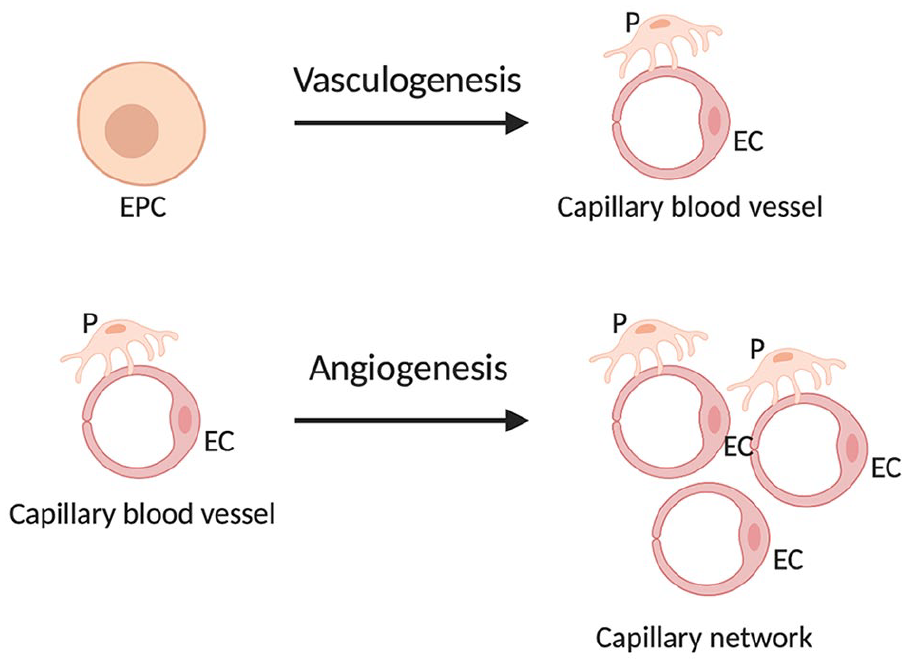
Mechanisms of the formation of new blood vessels: vasculogenesis begins after the initiation of gastrulation in the mammalian embryo whereby capillaries are formed from vascular progenitor cells; angiogenesis is the formation of new blood vessels from existing blood vessels.
Angiogenesis is the growth of blood vessels from existing vasculature. The endothelial cells at the venous end of the capillary are stimulated by growth factors to undergo five distinct processes: basement membrane digestion, migration, mitosis, basement membrane formation, and lumen formation. Angiogenesis is regulated under a fine balance of stimulation and inhibition. Under certain physiological (e.g. wound healing and menstruation), as well as pathological conditions including hypoxia and injury, growth factors are released in excess, thereby activating angiogenesis.10,11 Angiogenesis will continue until the pro-angiogenic stimulus is dampened or removed, and eventually the angiogenic inhibitors will be in excess, causing it to cease. The duration and intensity during which angiogenesis is ‘turned on’ determines whether the process is physiological or pathological. 12
The lymphatic system develops through a process known as lymphangiogenesis and primarily controls tissue fluid homeostasis, and serves as a trafficking route for immune cells. Hence, defects within the system will result in lymphedema formation and the compromise of the immune function. The lymphatic vessels develop from the endothelial cells that bud from the venous system and are mainly regulated by the VEGF-C/VEGF-D/VEGFR-3 signaling system. 13
Vascular morphogenesis is a well-regulated process through the interactions and effects of pro-angiogenic and antiangiogenic factors. The disruption in any components of the vasculogenesis and angiogenesis remodeling pathways could provide clues to the potential genes that have been mutated in inherited vascular malformation syndromes. The best characterized inherited vascular malformation syndromes include: type I and II hereditary lymphedema, hypotrichosis-lymphedema-telangiectasia, cutaneomucosal venous malformation, glomuvenous malformation (GVM), cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL), cerebral cavernous malformation, and hereditary hemorrhagic telangiectasia (HHT).
Molecular pathogenesis
The key signaling pathways involved in the pathogenesis of vascular malformations are summarized in Figure 2 and Table 2. Five key pathways have been identified to play a role in the pathogenesis of vascular malformations and are described as follows.
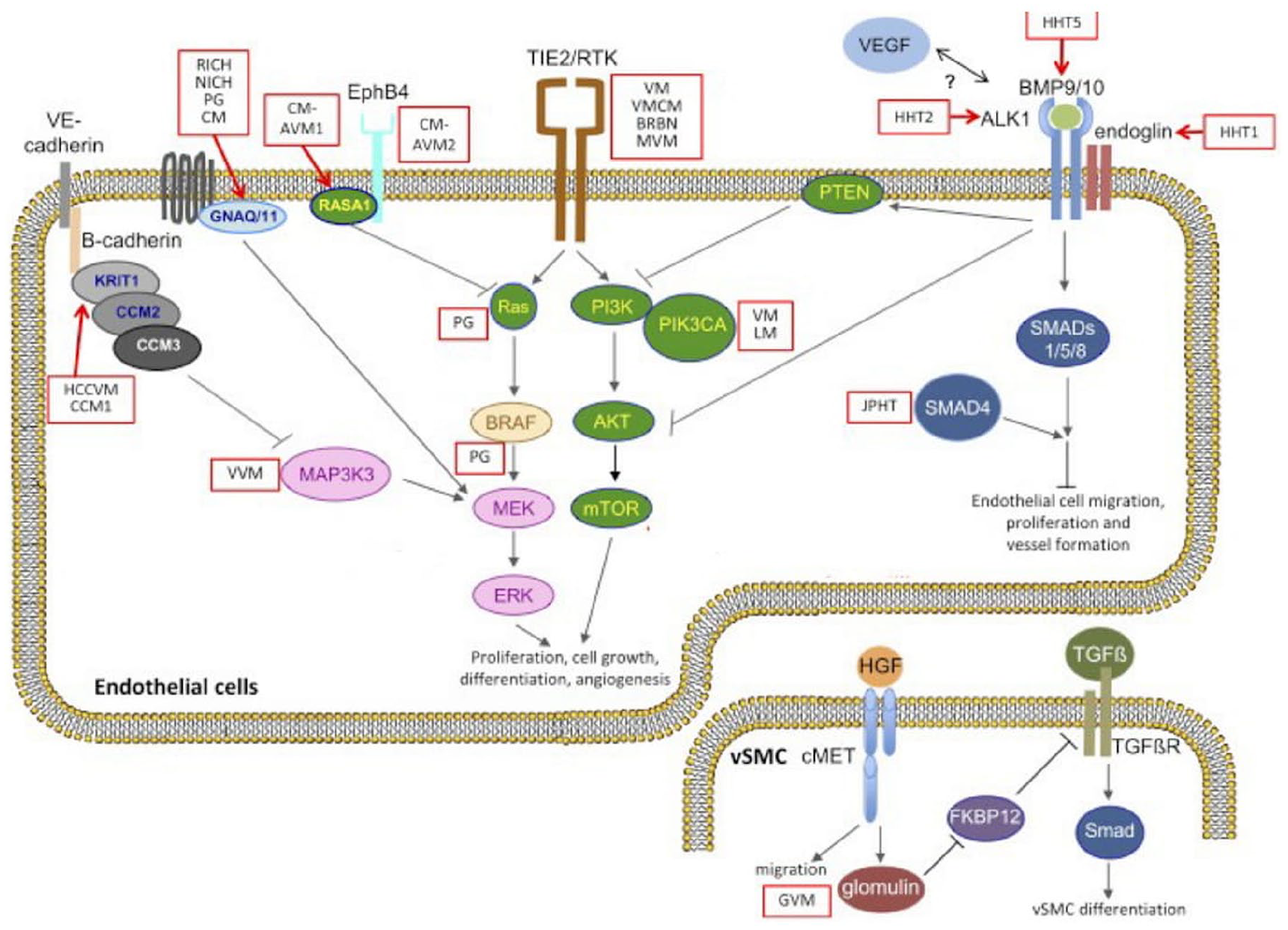
Diagram illustrating the mutations and signaling pathways involved in vascular anomalies.
An overview of vascular malformations with associated genetic cause.
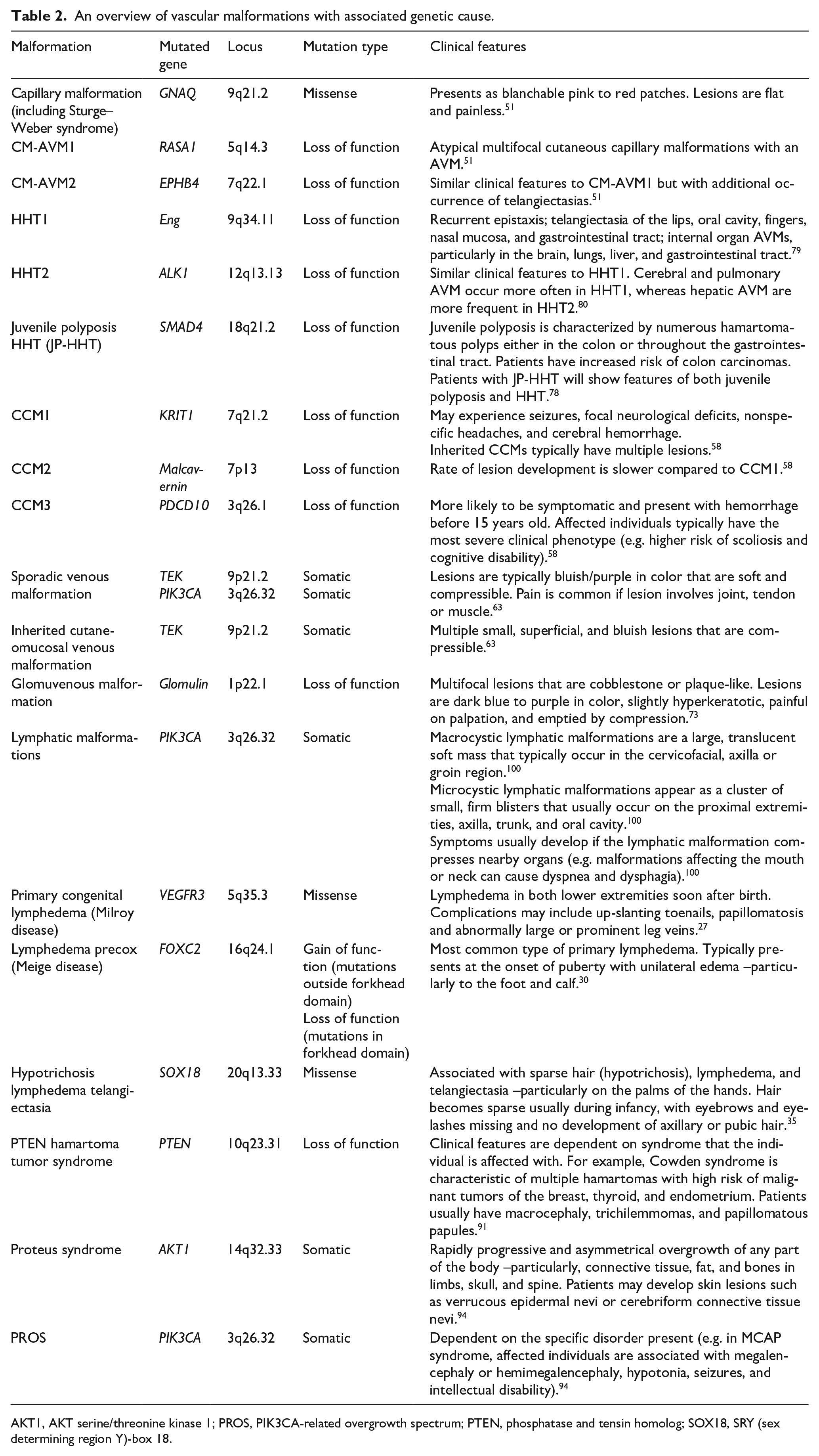
AKT1, AKT serine/threonine kinase 1; PROS, PIK3CA-related overgrowth spectrum; PTEN, phosphatase and tensin homolog; SOX18, SRY (sex determining region Y)-box 18.
ALK1, activin receptor-like kinase 1; AVM, arteriovenous malformation; CCM, cerebral cavernous malformations; CM-AVM, capillary malformation-arteriovenous malformation; Eng, endoglin; EPHB4, ephrin type-B receptor 4; FOXC2, forkhead box protein C2; GNAQ, guanine nucleotide-binding protein G(q) subunit alpha; HHT, hereditary hemorrhagic telangiectasia; KRIT1, Krev interaction trapped 1; PDCD10, programmed cell death protein 10; PIK3CA, phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha; RASA1, RAS p21 protein activator 1; SMAD4, homologs of the Drosophila protein, mothers against decapentaplegic (MAD) and the Caenorhabditis elegans protein Sma 4; TEK, tyrosine kinase, endothelial; VEGFR, vascular endothelial growth factor receptor.
Vascular endothelial growth factor (VEGF)
The VEGF signaling pathway plays a pivotal role in vasculogenesis, angiogenesis, and lymphangiogenesis. The VEGF family includes VEGF-A, VEGF-B, VEGF-C, VEGF-D, VEGF-E, and placental growth factor-1 and -2.14,15 Signaling is mediated by the binding to three membrane-bound tyrosine kinase receptors: VEGFR-1, VEGFR-2, and VEGFR-3. Both VEGFR-1 and VEGFR-2 are confined to endothelial cells during embryonic development.16,17 VEGFR-2 is exclusively localized on endothelial cells, hence playing an important role in endothelial cell differentiation and vasculogenesis.18,19 VEGFR-3 primarily regulates the development and regulation of the lymphatic system. 20 VEGF is the most potent direct-acting angiogenic protein,21,22 which mediates its response by increasing vascular permeability, and inducing endothelial cell proliferation, migration, survival, and secretion of matrix metalloproteinases. 23
Primary congenital lymphedema (Milroy disease) is caused by missense mutations in VEGFR-3,24,25 inhibiting the phosphorylation of the receptor and downstream signaling. 26 Milroy disease causes lower limb lymphedema that is present at birth or develops in infancy. Swelling is typically bilateral and can be associated with hydrocele and/or urethral abnormalities (males), prominent veins, up-slanting nails, hyperkeratosis, papillomatosis and/or cellulitis. 27 Management is aimed at reducing swelling and preventing complications, which typically involves conservative approaches that include the use of compression therapy and manual lymphatic drainage, while recurrent cases of infection may benefit from prophylactic antibiotics. 28
Primary lymphedema praecox (Meige disease) is caused by gain of function mutations in transcription factor FOXC2.28,29 It typically presents with unilateral lower limb lymphedema that rarely extends above the knee, which commonly occurs after puberty and predominantly in females. 30 Patients may develop cobblestone-like hyperkeratosis and cellulitis. 30 Its management follows the same approach as primary congenital lymphedema. FOXC2 is involved in lymphatic endothelial cell differentiation by regulating important endothelial target genes such as Ang-2, integrin β3, DLL4, and HEY2.31–33 In the lymphatic vessels, VEGF-C binds to its receptor VEGFR-3 and stabilizes FOXC2. In FOXC2-deficient lymphatic vessels, there has been demonstration of failed down-regulation of VEGFR-3 and its signaling. 34 Loss of FOXC2 leads to the overproduction of platelet derived growth factor subunit-B (PDGF-B) and increased recruitment of smooth muscle cells in the lymphatic walls. 26
Hypotrichosis lymphedema telangiectasia is caused by missense mutations in transcription factor SOX18. 35 It is characterized by the triad of sparse hair, lymphedema, and cutaneous telangiectasias. Sparse hair usually occurs during infancy with eyebrows and eyelashes missing and the lack of development of axillary or pubic hair. Lymphedema typically affects lower limbs, and telangiectasias are present on palms, soles, scalp, scrotum or legs. 35 Its management is directed towards the specific presenting symptoms, and primarily aimed at reducing swelling and preventing infection. The SOX18 expression can be detected in endothelial cells. It regulates prospero homeobox 1 (PROX1), which then controls the VEGFR-3 expression. PROX1 is an important regulator of lymphatic vessel differentiation, hence an early marker of lymphangiogenesis. 35 In endothelial cells, PROX1 interacts with MEF2C to regulate the adhesion molecule VCAM1 (vascular cell adhesion molecule 1), which plays a critical role in vascular and lymphatic endothelium. 26
Ras/Raf/MEK/ERK
The Ras/Raf/MEK/ERK pathway plays a prominent role in endothelial cell function.36,37 Targeted deletion of genes in this pathway are associated with vascular defects during embryogenesis.38,39 This pathway primarily functions by involving: (i) Ras recruiting and activating protein kinase Raf; (ii) Raf promoting MEK1/2 protein kinase and activation of ERK1/2; and (iii) activated ERK1/2 regulating different transcription factors. 40 Ras is a small GTP-binding protein that is an upstream molecule of several pathways such as MEK/ERK and PI3K/AKT. 41 Raf, a serine/threonine protein kinase, are the main effectors of Ras and upstream activators of the ERK pathway. 42 MEK1 and MEK2 are tyrosine and serine/threonine dual specificity protein kinases which catalyze the phosphorylation of ERK1/2. 43 ERK1/2 are members of the MAPK family that mediate cell proliferation and apoptosis. 44 The MEK5/ERK5 MAPK cascade has recently been implicated in embryonic vascular development and the maintenance of vascular integrity in mature blood vessels through knockout studies.45–47 In relation to the ERK1/2 MAPK, ERK5 activation is triggered by dual phosphorylation at a TEY consensus motif by MEK. 48 Missense mutations in GNAQ have been identified in capillary malformations. This protein encodes the guanine nucleotide binding protein G9q alpha subunit that hydrolyses GTP to GDP. This activates GTP-dependent signaling leading to the activation of the mitogen-activated protein kinase (MAPK) signaling. 49
Capillary malformations-arteriovenous malformations (CM-AVM) is an autosomal dominant condition caused by loss of function mutations in RASA1.26,50 The hallmark of this disorder is the disorganized distribution of multiple atypical cutaneous capillary malformations, with associated high-flow lesions. 50 Capillary malformations are typically small, round-to-oval in shape, and pink-red in color. These malformations are characteristically surrounded by a blanched halo with an associated steal phenomenon and increased blood flow which can be detected on a Doppler ultrasound. 51 Patients who are symptomatic or with suspected CM-AVM should be investigated with a magnetic resonance imaging (MRI) for intracranial and spinal AVMs. 52 RASA1 codes for RAS p21 protein activator 1 (p120-Ras-GAP), which negatively regulates Ras/MAPK/ERK signaling. Mutations in this gene cause the loss of function of GTPase activity of Ras, hence overstimulation resulting in aberrant cell growth, differentiation, proliferation, and endothelial cell network organization.53,54 A sub-entity, CM-AVM2, is caused by the loss of function mutations in EphB4, 55 which is a transmembrane receptor on the surface of venous endothelial cells. 56 Ephrin-B2 is a protein found on arterial endothelial cells that binds to its receptor EphB4. 57 EphB4 is involved in the inhibition of the Ras/Raf/MEK/ERK signaling through interactions with p120-Ras-GAP. 58
Cerebral cavernous malformations (CCM) is caused by loss of function mutations in three genes: (i) Krev interaction trapped 1 (KRIT1 or CCM1); (ii) cerebral malcavernin or CCM2; and (iii) programmed cell death protein 10 (PDCD10 or CCM3). 58 CCMs are largely located within the central nervous system. Approximately 25% of individuals with CCMs never experience any related medical problems. 54 However, cerebral hemorrhage from CCMs may cause seizures and neurological deficits such as muscle weakness, loss of sensation, and paralysis. 58 Accessible lesions can be surgically removed or embolized. Medical treatment can be used to manage associated symptoms such as the use of anti-epileptics to prevent seizures. 59 CCM1 is involved in regulating endothelial cell-cell junctions through Delta-Notch signaling, leading to the activation of AKT and decreased ERK activity. 60 CCM1 interacts with CCM2 and CCM3 to form a complex that regulates MAP3K3 and GTPase RAC1 function. 61 The loss of this CCM complex function activates MAP3K3 signaling and its target genes KLF2, KLF4, RHO, and ADAMTS. 62 Moreover, depletion of CDC42 (cell division cycle 42) in brain endothelial cells has been shown to elicit increased MEKK3-MEK5-ERK5 signaling and subsequent overexpression of KLF2 and KLF4. CDC42 and KLF4 are downstream from MEKK3, which is upstream from ERK5, hence are examples of emerging molecular targets for CCM and other vascular malformations related to this pathway.63,64
Angiopoietin-TIE2
Sporadic and cutaneomucosal venous malformations are characterized by small (< 2 cm in diameter), multifocal, soft, usually compressible, and bluish-purple venous lesions involving the skin and mucous membranes (Figure 3). Larger lesions may infiltrate the underlying muscle and joints – causing pain and sometimes calcifications. 65 Its management is often based upon the location and extent of the malformations, consisting of the use of compression garments, analgesia, and sclerotherapy as first-line intervention. 66
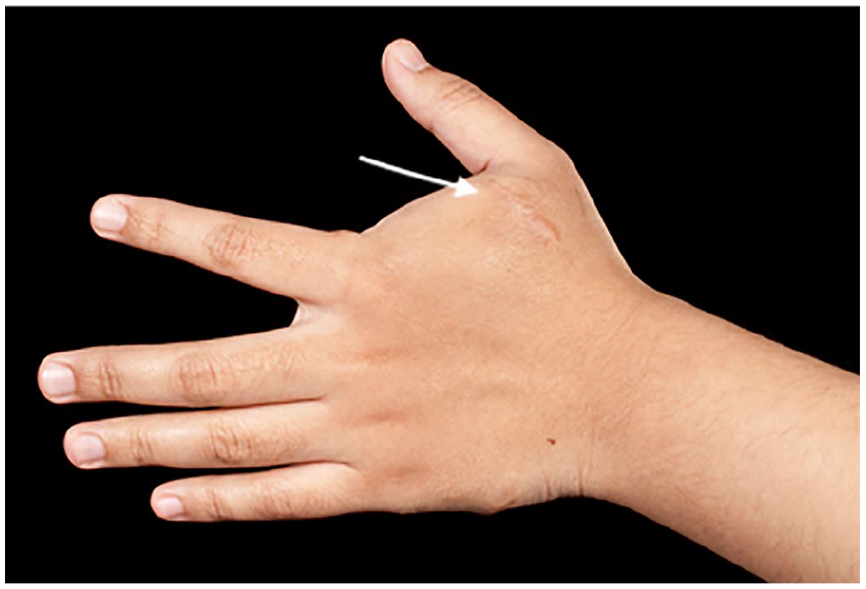
Venous malformation (arrow) localized to the hand. These are typically bluish, soft, and compressible lesions.
Sporadic and cutaneomucosal venous malformations are caused by somatic mutations in TIE2, which is an endothelial cell-specific tyrosine kinase receptor that binds angiopoietins and encodes for the TEK gene. 67 TIE2 has three known ligands: angiopoietin 1 (Ang1), angiopoietin 2 (Ang2), and angiopoietin 4 (Ang4). The angiopoietin and Tie families play an important role in the later stages of vascular development and in adult vasculature by controlling remodeling and stabilization of vessels. Ang1 is required for the maturation and stability of newly formed vessels but Ang2 acts as an antagonist of Ang1/TIE by interfering with the stabilizing effects of Ang1 by allowing blood vessels to respond to pro-angiogenic factors and undergo growth and remodeling.68,69 Ang1 activates TIE2, resulting in receptor phosphorylation, thereby stimulating many intracellular pathways – in particular the PI3K/AKT/mTOR pathway, which will be discussed shortly. 26
Transforming growth factor beta (TGF-β)
TGF-β is a multifunctional cytokine that plays a key role during embryogenesis. TGF-β signaling is through two classes of cell surface serine/threonine kinase receptors, type I and II, which recognize TGF-β family ligands. Upon ligand binding, the type II receptor phosphorylates the type I receptor, thereby providing a binding site for intracellular effectors of the pathways, the SMADs. 70 The receptor-regulated SMADs (R-SMADs) become phosphorylated by the type I receptor and form a heteromeric complex with one common-SMAD (co-SMAD), SMAD4. TGF-β is generally split into two pathways, with TGF-β/activin leading to phosphorylation of SMAD2 and SMAD3, and bone morphogenetic protein (BMP) leading to phosphorylation of SMAD1, SMAD5, and SMAD9. 71 TGF-β regulates endothelial cells by activating activin receptor-like kinase (ALK) 1 and ALK5. 72 TGF-β/ALK1 signaling induces SMAD1/5 activation, which stimulates endothelial cell migration, proliferation, and tube formation. 73 Meanwhile, TGF-β/ALK5 signaling induces SMAD2/3 phosphorylation and blocks angiogenesis by inhibiting endothelial cell proliferation, tube formation, and migration.72,73
GVM is caused by the loss of function mutations in glomulin.26,74,75 GVMs are usually pink to purple-bluish lesions that are located on the extremities, affecting the skin and rarely the mucosa. They appear as nodular and multifocal raised with a cobblestone-like appearance, except for the rare plaque-like lesions. 75 Lesions are not compressible, but painful on palpation. 71 Surgical excision is the treatment of choice in isolated lesions while sclerotherapy is effective in multiple lesions.76,77 Glomulin is involved in vascular smooth muscle cell differentiation 76 through TGF-β signaling. The binding of FK506 binding protein 12 (FKBP12) to the TGF-β type I receptor causes inhibition of the TGF-β signaling. 26 Glomulin interacts with FKBP12 by forming a complex, which has been shown to be inhibited by rapamycin. This may suggest glomulin has a role in the mammalian target of the rapamycin (mTOR) signaling pathway. 78 Mutations in this gene will therefore enable FKBP12 to bind to its receptor, hence inhibit TGF-β signaling. 26 Different SMAD signaling cascades are then recruited and activated depending on which type I receptor is activated. SMADs comprise a family of structurally similar proteins that are the main signal transducers for receptors of the TGF-β. In endothelial cells, TGF-β signals through ALK1.
In HHT, loss of function mutations in three genes has been identified. HHT1 is caused by the loss of function mutations in endoglin (ENG). 79 HHT2 is caused by mutations in the ALK1 gene. 80 Juvenile polyposis HHT is caused by the loss of function mutations in MADH4, which encodes the downstream effector SMAD4. 80 HHT is manifested by spontaneous and recurrent epistaxis, mucocutaneous telangiectasias, and visceral AVMs. 81 These visceral AVMs are usually asymptomatic but may lead to serious hemorrhagic complications. Pulmonary AVMs may present with dyspnea on exertion and hypoxemia. Paradoxical emboli can lead to serious complications such as brain abscesses, strokes, transient ischemic attacks, and hemorrhagic rupture. Cerebral AVMs may bleed causing hemorrhagic stroke. Hepatic AVMs may manifest as high-output cardiac failure, portal hypertension or pulmonary hypertension. Again, its management is primarily aimed at preventing and treating the complications such as hemorrhage and anemia, and screening for new or worsening AVMs. 82 Signaling through ALK1 induces and inhibits endothelial cell migration and proliferation,83,84 which is modulated by ENG. 85 Recently, BMP9 and BMP10 have been shown to be associated with the pathogenesis of HHT. Binding of BMP9 and BMP10 to ALK1 or ENG inhibit endothelial cell proliferation and migration. 86 ENG increases BMP9/BMP10/Alk1 signaling, resulting in the activation of receptor-regulated SMAD1/5/8 and co-SMAD, SMAD4. This suppresses endothelial cell migration and proliferation, thereby maintaining a quiescent endothelium. 87
PI3K/AKT/mTOR
The PI3K/AKT/mTOR pathway plays an important role in cellular proliferation, adhesion, migration, invasion, metabolism, and survival. 88 PI3K activation occurs via Ras mutation, loss of PTEN (phosphatase and tensin homolog) or by increased expression of growth factors. The activation is through the PI3K family, which leads to the phosphorylation of phosphatidylinositol-4,5-biphosphate (PIP2) to phosphatidylinositol-3,4,5-triphosphate (PIP3). PIP3 is a potent signaling molecule that recruits and regulates numerous downstream effectors, particularly the AKT, which controls protein synthesis and cell growth. 89 PTEN is a negative regulator of this pathway by opposing PI3K activity, thereby inhibiting AKT activation. 90
PTEN hamartoma tumor syndrome is a spectrum of disorders associated with the loss of function mutations in PTEN. PIP3 is converted to PIPs by PTEN. The mutations in this gene will result in constitutive activation of PI3K/AKT/mTOR signaling.91,92 This disorder consists of Cowden syndrome, Bannayan–Riley syndrome, Proteus syndrome, and Proteus-like syndrome. The disease characteristics are dependent on the specific disorder. Cowden disease is characterized by multiple benign growths, or hamartomas with an increased risk of both benign and malignant tumors. The most common phenotypic features include trichilemmomas, papillomatous papules, and acral and plantar keratoses. Patients are at increased risk of certain types of cancer, including breast, thyroid, and renal cell carcinoma. 93 Bannayan–Riley syndrome is characterized by macrocephaly, hamartomatous intestinal polyps, pigmented macules of the penis, and neurodevelopmental disorders including intellectual disability and autism. 94 Proteus syndrome is caused by somatic mutations in the AKT1 gene 95 resulting in abnormal growth and division. It is characterized by rapidly progressing overgrowth, typically asymmetric in a mosaic distribution, of the bones, skin, and other tissues that usually manifest between 6 months and 2 years. 96 Meanwhile, there are individuals with Proteus-like syndrome who demonstrate many features associated with Proteus syndrome, but do not meet the diagnostic criteria. 97 Cancer surveillance is the key element of its management due to the increased risk of malignancy associated with this disease. Surveillance programs may include mammography and breast MRI, and thyroid ultrasound.64,92
PIK3CA-related overgrowth spectrum (PROS) is associated with somatic mutations in the PIK3CA gene 95 resulting in abnormal activation of PI3K-AKT signaling, leading to an increase in cellular proliferation. PROS encompasses overgrowth syndromes that include: megalencephaly-capillary malformation syndrome; dysplastic megalencephaly; congenital lipomatous asymmetric overgrowth of the trunk; lymphatic, capillary, venous, and combined-type vascular malformation; congenital lipomatous overgrowth; vascular malformations; epidermal nevi, and spinal/skeletal anomalies (CLOVES) syndrome; hemihyperplasia-multiple lipomatosis; and fibroadipose overgrowth, and Klippel–Trenaunay syndrome (KTS) (Figure 4). Presentation is dependent on the clinical entities of the syndromes. 96 For example, KTS is characterized by a triad of port-wine stain, abnormal growth of soft tissues and bones, and venous malformations (Figure 5). The overgrowth of bones and soft tissues typically affects a single lower limb, causing pain and limb length discrepancy. Venous malformations can involve the pelvic or abdominal organs, causing bleeding from the rectum, vagina or bladder. Extensive venous abnormalities can lead to hematological consumptive complications such as localized intravascular coagulopathy (LIC). Anticoagulation (e.g. with low molecular weight heparin) can be used to treat the pain associated with LIC to prevent potential progression to disseminated intravascular coagulopathy (DIC).98,99 In half of the venous malformations that are not associated with TIE2 mutations, PIK3CA mutations have been identified.50,97 This causes the disruption of the endothelial cell monolayer, loss of extracellular matrix fibronectin, and downregulation of ANGPT2 and PDGF-B expression.54,56 Several lymphatic malformations have also been shown to be caused by PIK3CA mutations.100,101
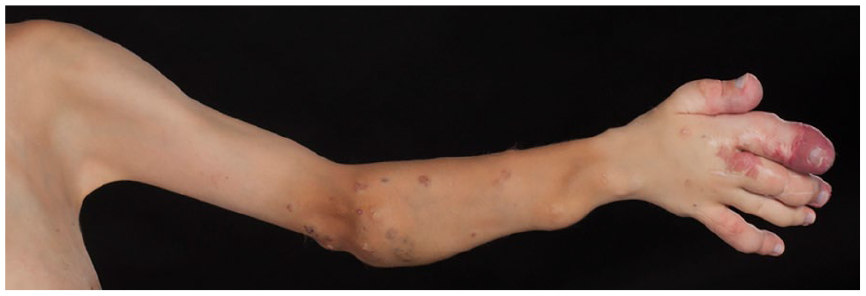
PIK3CA-related overgrowth spectrum involving the left upper limb demonstrating hand overgrowth with associated capillary malformations.
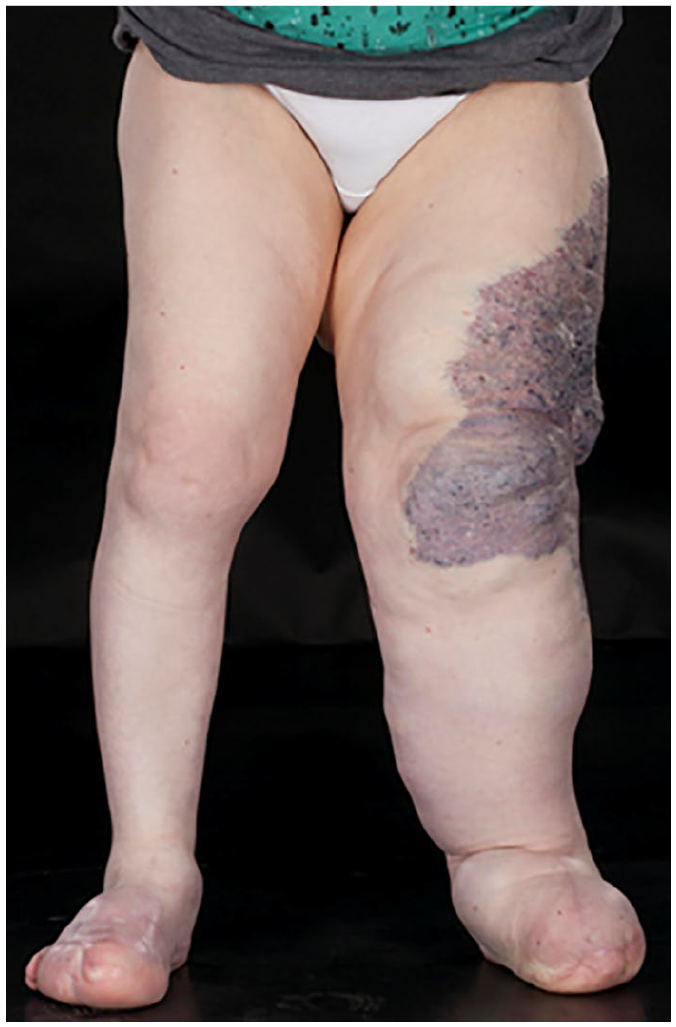
Klippel–Trenaunay syndrome involving the left lower limb demonstrating a large port-wine stain and marked hypertrophy.
Emerging medical treatments that are based on molecular pathways of vascular malformation pathogenesis
Vascular malformations should be managed using a multidisciplinary approach. Lidsky and colleagues showed that implementation of diagnostic and therapeutic algorithms in a multidisciplinary setting resulted in favorable outcomes with an acceptable complication rate in patients with vascular malformations. 102 The diagnosis of a vascular malformation is often clinical, and typically confirmed with radiological imaging. Low-flow and high-flow lesions can often be differentiated clinically, or with the help of a hand-held Doppler and/or duplex ultrasonography. On duplex ultrasonography, the low-flow lesions typically demonstrate heterogenous echotexture with a monophasic flow on Doppler. Macrocystic lymphatic malformations (Figure 6) show enlarged cystic spaces, whereas microcystic lymphatic malformations demonstrate a hyperechoic echotexture but with no Doppler flow. 103 In contrast, AVM will reveal a high flow and shunting within a region of high vessel density. 103 Cross-sectional imaging, including MRI or, rarely, computed tomography (CT), is then used to confirm and characterize the lesion, particularly anatomically, and is essential for treatment planning. 104 For high-flow lesions, MRI ± 4D time-resolved magnetic resonance angiography may be indicated for accurate assessment of feeding and draining vessels, which helps in treatment planning. 105 Lesions with unusual clinical and imaging features should be evaluated with a tissue biopsy to exclude vascular tumors (e.g. sarcoma) (Figure 7). 106
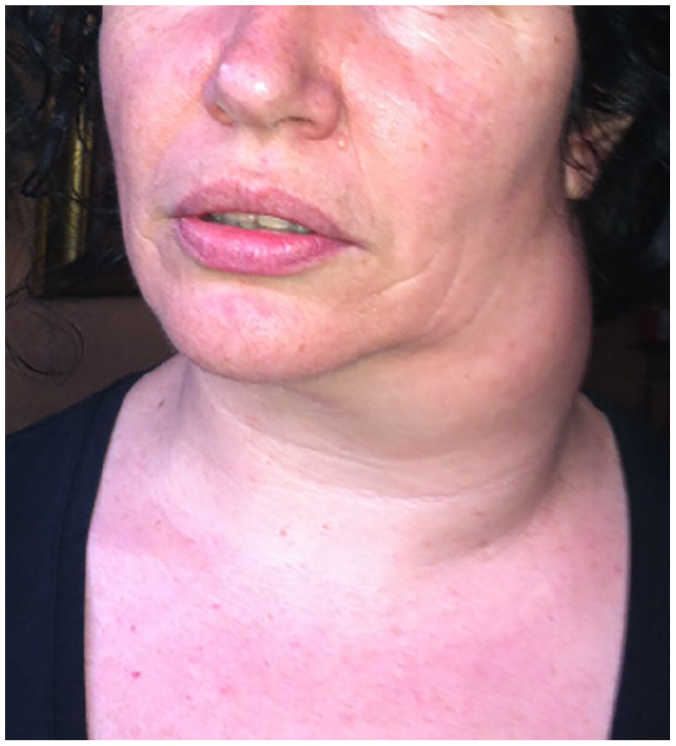
Macrocystic lymphatic malformation on the side of the neck (cystic hygroma).
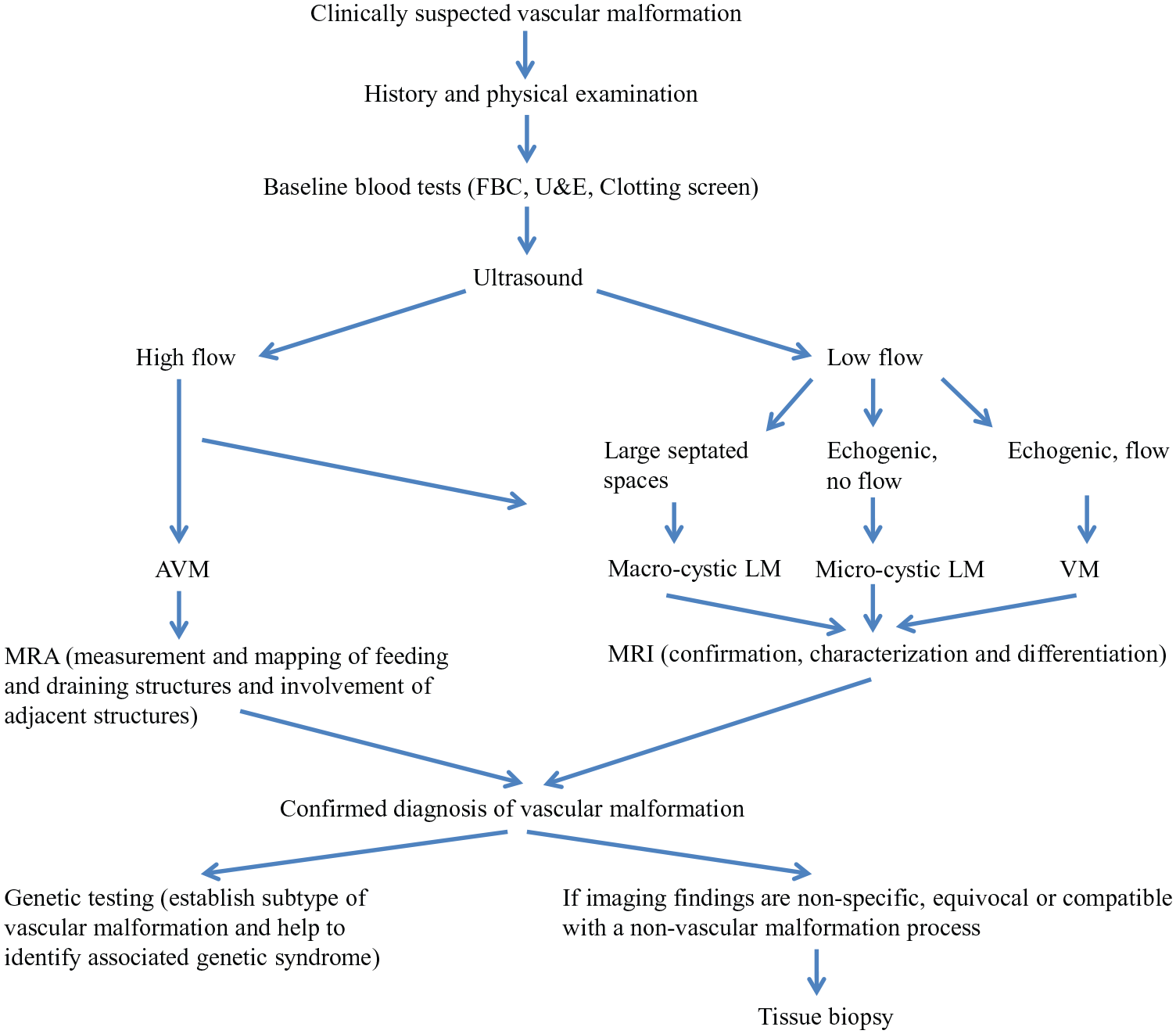
A diagnostic pathway for vascular malformations.
The current management options for vascular malformations are largely conservative and supportive with interventions including embolo-sclerotherapy and surgery as necessitated by symptoms. General measures include physiotherapy, graduated compression hosiery, support and education, and psychological counselling. Endovascular therapy is the main interventional therapeutic tool in the management of vascular malformations and is considered a relatively safe and effective treatment. 107 However, these interventions are invasive with associated risks of complications. Furthermore, current management of vascular malformations is limited due to the restricted understanding of the molecular pathogenesis of vascular malformations and, consequently, the lack of specific target therapy. This is further complicated by the heterogeneity of vascular malformations as well as the diverse and often traumatized patient groups. New and existing classification systems for vascular malformations incorporating the aberrant signaling pathways involved, such as in Table 3, may help to predict prognosis and improve the overall management of these patients (Figure 8).
Proposed new classification of vascular malformations based on signaling pathways.
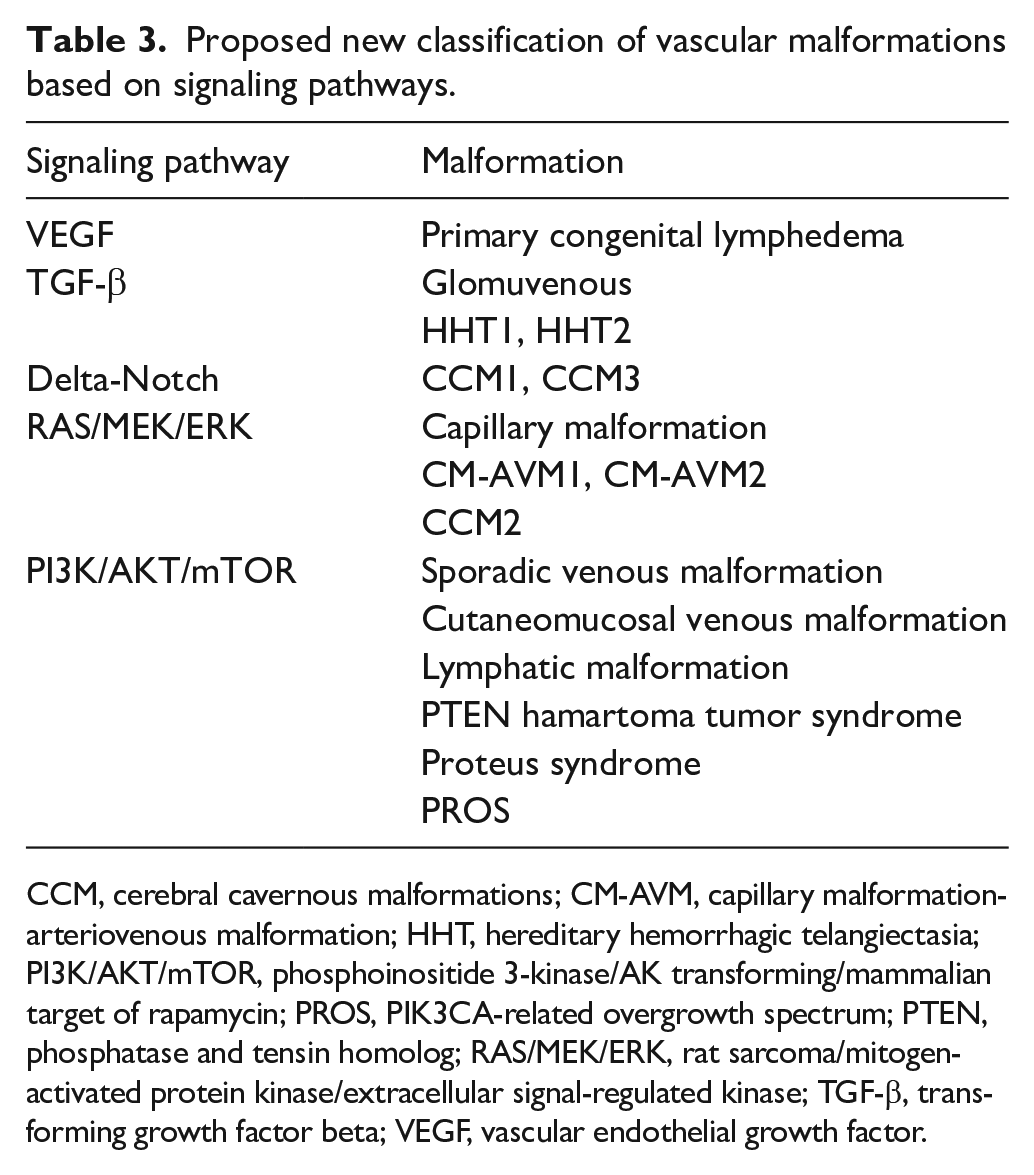
CCM, cerebral cavernous malformations; CM-AVM, capillary malformation-arteriovenous malformation; HHT, hereditary hemorrhagic telangiectasia; PI3K/AKT/mTOR, phosphoinositide 3-kinase/AK transforming/mammalian target of rapamycin; PROS, PIK3CA-related overgrowth spectrum; PTEN, phosphatase and tensin homolog; RAS/MEK/ERK, rat sarcoma/mitogen-activated protein kinase/extracellular signal-regulated kinase; TGF-β, transforming growth factor beta; VEGF, vascular endothelial growth factor.
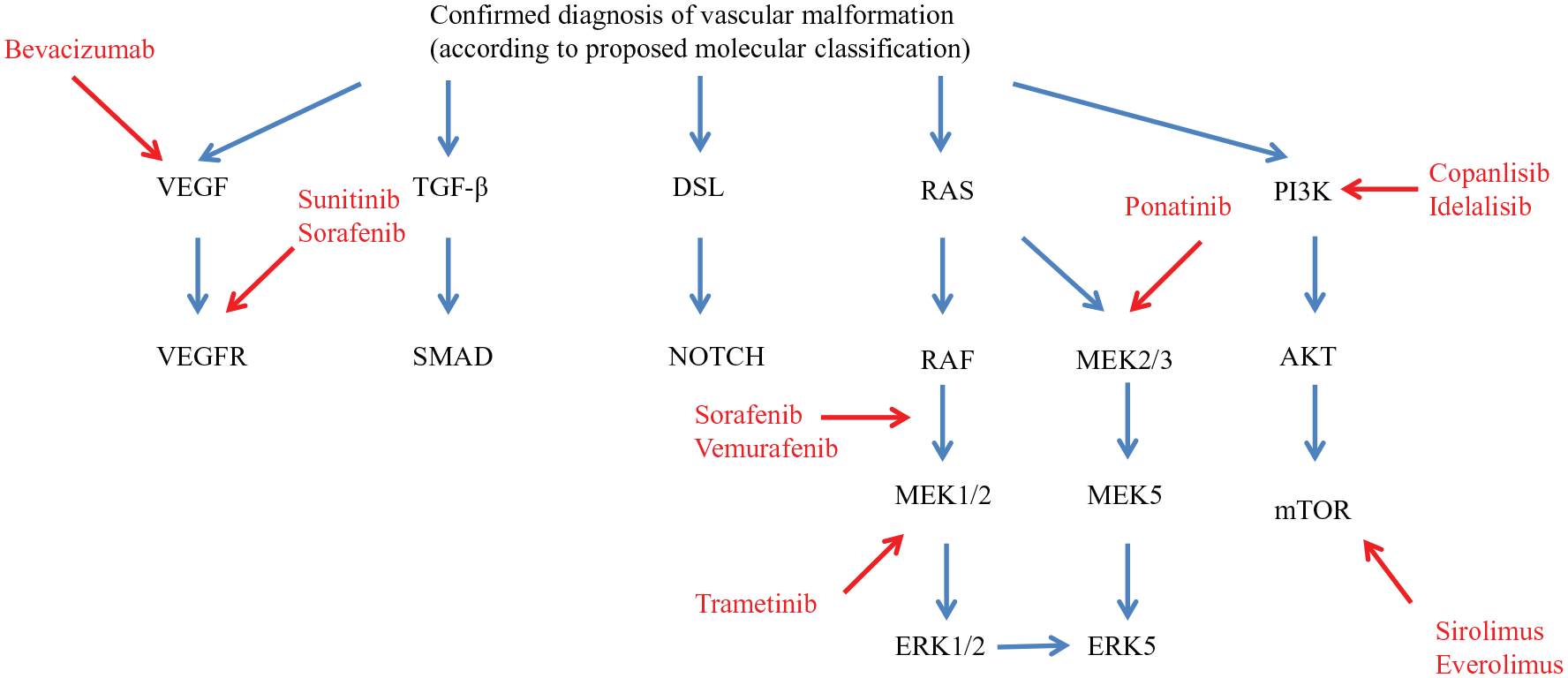
Potential therapeutic targets for vascular malformations based on current understanding and proposed molecular classification (see Table 3).
Recent improvement in the understanding of the molecular pathogenesis of vascular malformations has led to the deployment of several targeted medical therapies for vascular malformations with specific aberrant signaling pathways. Sirolimus, an mTOR inhibitor, is used mainly as an immunosuppressive agent to prevent organ rejection. However, several trials have shown its efficacy in extensive and/or complex low-flow vascular malformations that were refractory to standard treatments.108–110 Thalidomide and bevacizumab (Avastin) are antiangiogenic agents that are used in various cancers where VEGF plays an important role. Both these drugs have been used in the treatment of complications associated with HHT, such as severe epistaxis, gastrointestinal bleeding, and high-output cardiac failure secondary to hepatic arteriovenous malformations.111–115 Early studies report the utility of thalidomide as a medical adjunct in the management of severe and refractory high-flow AVM. 116 Despite increasing reports of medical treatments for the management of vascular anomalies, results of efficacy and safety are based largely on case reports/small case-series and clearly much further work with larger trials is needed. However, our review aims to highlight the potential of these molecular pathways as future therapeutic targets.
Classifying vascular malformations based on their molecular pathways
Molecular biology has enabled a better understanding of the pathogenesis of vascular anomalies. This has provided insights into molecular mechanisms underlying vascular morphogenesis, growth, and development. Vascular malformations have been shown to be strongly linked to endothelial cell signaling pathways – particularly Ras/Raf/MEK/ERK and PI3K/AKT/mTOR. 117 These pathways have been shown to cause various cancers and hence the use of anticancer drugs may be beneficial for vascular malformation treatment. An example is sirolimus, an mTOR inhibitor which has shown beneficial effects in the treatment of lesions associated with the PI3K/AKT/mTOR pathway.108,109
The ISSVA classification provides a systematic approach to vascular lesions that corresponds with clinical history, presentation, and disease manifestation. 118 The limitation is that vascular anomalies acquired after infancy, such as von Hippel–Lindau syndrome, cannot be classified under a current ISSVA category. 119 The classification system is based on histopathological aspects of vascular anomalies; this presents difficulties in categorizing lesions with limited biopsy results such as rapidly involuting congenital hemangioma. Multiple separate disease entities as described in the ISSVA classification have been shown to share a common gene signaling pathway mutation (e.g. PROS and KTS) and appear to respond positively to sirolimus therapy, suggesting a common etiological heritage. In classifying these disparate lesions by their common molecular signaling pathway mutations, simplification of classification of direct therapeutic relevance becomes possible. The process of development of the classification, ultimately to include all vascular malformations, can only become more robust as further active agents are discovered and assessed. With this in mind, and the rapid discovery of underlying genes involved, a molecular classification may be more appropriate, or at least complementary in the challenging management of vascular malformations.
A molecular classification enables a better understanding of the pathophysiology of vascular anomalies. The identification of the signaling pathway involved can provide a targeted therapy approach and has potential to become a prognostic/predictive test. The limitations of a molecular classification are the ability to consistently assign a molecular class to new cases of vascular anomalies and also uncertainty of the number of molecular classes there may be. Further research will be required to generate human data through analysis of gene-expression profiles to gain insight into their function with potential new drugs. This can be achieved with the available genetic tests that are validated clinically for vascular anomalies. These tests are based on a targeted next generation sequencing approach, which will help identify genotype–phenotype correlations and possible new therapeutic targets for certain gene mutations. 120
Conclusion
Several molecular signaling pathways with potential therapeutic targets have been identified and demonstrated to contribute to the development of various vascular anomalies. Compared to current classification systems, classifying vascular malformations based on their molecular pathogenesis is likely to improve diagnostic accuracy and relevance in terms of determining the underlying nature of the condition and their potential therapeutic target. A molecular classification of vascular anomalies as proposed, is clearly needed to improve and complement the current classification systems.
Footnotes
Acknowledgements
We thank Nicholas Evans for helping us obtain the clinical figures (Figs 3-6), and we thank the patients for granting permission to publish their photographs. This study was supported by researchers at the National Institute for Health Research University College London Hospitals Biomedical Research Centre.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Chung Sim Lim receives funding from the National Institute for Health Research, Biomedical Research Centre, University College London Hospitals, United Kingdom.