
Abstract
Critical limb ischemia (CLI) is associated with skeletal muscle damage. However, the pathophysiology of the muscle damage is poorly understood. Toll-like receptors (TLR) have been attributed to play a role in ischemia-induced tissue damage but their role in skeletal muscle damage in CLI is unknown. TLR2 and TLR6 expression was found to be upregulated in skeletal muscle of patients with CLI. In vitro, ischemia led to upregulation of TLR2 and TLR6 by myotubes, and activation of the downstream TLR signaling pathway. Ischemia-induced activation of the TLR signaling pathway led to secretion of the pro-inflammatory cytokine interleukin-6 and muscle apoptosis, which were abrogated by neutralising TLR2 and TLR6 antibodies. Our study demonstrates that TLR2 and TLR6 are upregulated in ischemic muscle and play a role in ischemia-induced muscle damage. Thus, manipulating the TLR pathway locally may be of potential therapeutic benefit.
Keywords
Introduction
Peripheral artery disease (PAD) is a major cause of morbidity and mortality and is caused by the accumulation of atherosclerotic plaques within the lower limb arteries. Critical limb ischemia (CLI) results from progression of this disease leading to chronic muscle ischemia with tissue inflammation, necrosis, and apoptosis. 1 Patients with CLI present with rest pain with or without ulcers or gangrene. At this stage, limb salvage requires surgical or endovascular attempts to improve blood flow; however, in some patients, revascularisation is not possible. An increasing number of studies show that even successful revascularisation does not always lead to improvements in symptoms, functional status or quality of life.2,3 Indeed, 34% of patients with CLI undergo major limb amputation within 12 months. 4 The lack of improvement in symptoms in patients despite revascularisation is due in part to a myopathy5–8 within the ischemic muscle. The pathophysiology of skeletal muscle damage following ischemia is unclear, but a number of detrimental physiological and biochemical changes have been identified. However, the mechanism of how hypoxia/ischemia activates the damaging pathways in muscle are yet to be elucidated.
Toll-like receptors (TLRs) are pathogen recognition receptors (PRR) of the innate immune system. They are type 1 membrane glycoproteins that contain an external ligand-binding leucine-rich repeat (LRR) motif and a cytoplasmic signaling domain that is similar to the interleukin-1 (IL-1) receptor and thus termed the Toll/IL-1 receptor (TIR) domain. So far, 11 TLRs (TLR 1–11) are thought to be functional in humans, and despite their documented role in innate immunity they are widely expressed by both immune and non-immune cells, including skeletal muscle. 9 TLRs recognise and are activated by pathogen-associated molecular patterns (PAMPs), which are microbial molecules that alert the host cell of the presence of invading organisms, such as bacteria and viruses. In addition to PAMPs, TLRs are also stimulated by host-derived molecules 10 that are released during tissue injury. Endogenous ligands, such as high mobility group box 1 protein (HMGB-1), 11 heat shock proteins (HSPs), and hyaluronan, 12 have been implicated in ischemia-induced organ damage.
Ligand binding by PAMPs or endogenous molecules leads to TLR dimerisation, which contributes to ligand specificity. All TLRs form homodimers except TLR1, 2 and 6: TLR2 can heterodimerise with TLR1 or TLR6 13 depending on the stimulating ligand. Exogenous or endogenous ligand stimulation of the TLR signaling pathways, of which there are two distinct types, leads to activation of transcription factors, such as nuclear factor κB (NF-κB), activator protein 1 (AP-1), and the interferon regulatory factors. This in turn leads to upregulation of pro-inflammatory cytokines, such as tumour necrosis factor α (TNF-α), interleukins 1 and 6 (IL-1 and IL-6), and type 1 interferons (IFN). Further, TLR activation has been reported to directly cause apoptosis via Fas-associated death domain protein (FADD) and caspase 8. 14
Signal transduction following TLR stimulation is dependent upon the TIR domain binding to an adapter protein. There are four adapter proteins that are involved: myeloid differentiation primary response protein 88 (MyD88), TIR domain-containing adapter protein (TIRAP/Mal), TIR domain-containing adapter-inducing IFNβ (TRIF), and TRIF-related adapter molecule (TRAM). 15 The two distinct TLR signaling pathways are the MyD88 dependent and the MyD88 independent pathways, whose main adapter proteins are MyD88 and TRIF, respectively. All TLRs signal via the MyD88 dependent pathway apart from TLR3, which signals via the MyD88 independent pathway, whilst TLR4 can signal via either signaling mechanism. The subsequent propagation of the signaling pathway involves the interleukin-1 receptor-associated kinase (IRAK) and MAP kinase family. In the TLR signaling pathway, the quiescent NF-κB is sequestered by the inhibitor of NF-κB (IκB) protein. Following TLR stimulation and signal transduction, IκB protein is phosphorylated and polyubiquitylated, which releases NF-κB to translocate to the nucleus and activate gene transcription.
There is emerging evidence suggesting that TLRs play an important role in ischemia and ischemia/reperfusion-induced tissue damage in the kidneys, liver, central nervous, and cardiovascular systems. 16 In particular, TLR2 and TLR4 activation has been strongly associated with ischemia-induced organ damage.17,18 TLR2 and TLR4 knockout mice have been shown to be protected against ischemia-induced liver, 19 renal, 20 and myocardial 21 injury. Further studies have shown that this protective effect is associated with a reduction in pro-inflammatory cytokine expression 22 and NF-κB nuclear translocation. 21 Despite this strong association between TLRs and ischemia-induced pathogenesis, very little work has focused on the role that TLRs play in skeletal muscle damage in CLI.
Current treatments for CLI are aimed at improving blood flow. However, there is significant evidence to suggest that this does not always help improve patient symptoms. We have therefore taken a novel approach of studying the affected end organ (i.e. the skeletal muscle itself) in order to understand whether TLR2 and TLR6 play a role in the ischemia-induced muscle damage. A better understanding of this pathological process may identify potential therapeutic targets that may be used in conjunction with revascularisation.
Methods
Human gastrocnemius muscle biopsy
Open gastrocnemius muscle biopsies were obtained from patients undergoing perigenicular amputation for CLI, and non-ischemic control samples were obtained from patients undergoing great saphenous vein harvesting for coronary artery bypass grafting. Twelve patients from each group were biopsied. The biopsy was taken as an excisional biopsy rather than using a biopsy needle. Thus, the sample size of the biopsy is approximately 4 × 4 mm. At least three biopsies were taken from each patient with PAD while one sample per patient was taken from the control patients. The sample is taken from the medial head of the gastrocnemius muscle and any areas with macroscopic necrosis were avoided. To ensure that none of the control patients had flow-limiting PAD, all patients with an ankle–brachial index (ABI) of < 0.9 were excluded from the control group. In addition, any patient with an infected ulcer or trauma was also excluded from the sample collection. There was no significant difference in the age-related risk factors between the two groups (Table 1). We have previously shown that hypoxia-inducible factor 1α, a marker of hypoxia, is elevated in muscle samples from patients with CLI compared to controls.
23
The same technique was used to harvest the muscle samples in both groups and the tissue was snap frozen in liquid nitrogen and stored at −80
Patient characteristics, including risk factors for atherosclerotic disease.
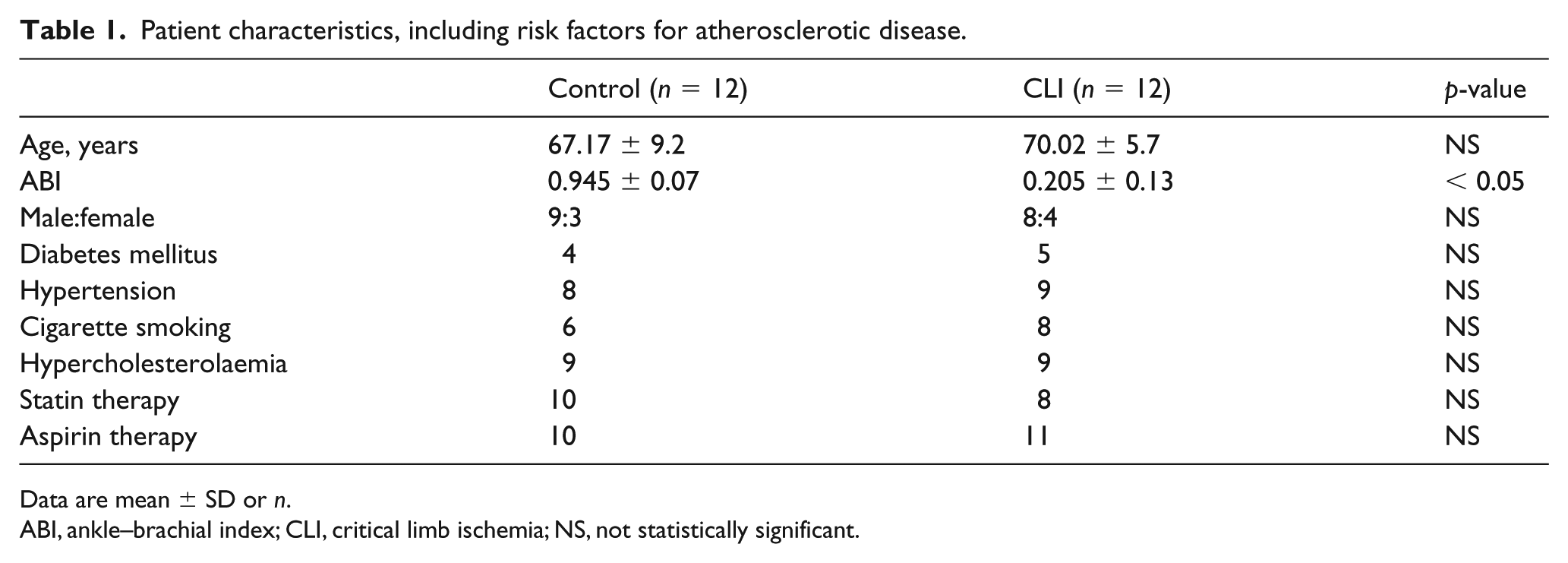
Data are mean ± SD or n.
ABI, ankle–brachial index; CLI, critical limb ischemia; NS, not statistically significant.
Cell cultures
The C2C12 mouse myoblast cell line (European Collection of Cell Cultures (ECACC), no. 91031101, passage no. 13, Salisbury, UK) was plated onto 12-well culture plates (BD Falcon™, cat. no. 351143, Flintshire, UK) or 8-well chamber slides (BD Falcon, cat. no. 354108) and cultured in Dulbecco’s modified Eagle’s medium (DMEM; Gibco, Paisley, UK) supplemented with 10% fetal calf serum, penicillin (100 U/mL), streptomycin (100 µg/mL), and amphotericin B (25 µg/mL) (all from Gibco). At 80% confluence, cells were switched to differentiation medium containing DMEM with 2% horse serum penicillin (100 U/mL), streptomycin (100 µg/mL), and amphotericin B (25 µg/mL) (all from Gibco). Differentiation was confirmed by changes in morphology (online supplementary Figure 1), as well as changes in the expression of myogenin protein. Myogenin is preferentially expressed in skeletal myotubes but not myoblasts. 23 Following differentiation, usually at day 7–8, myotubes were exposed to simulated ischemia or normoxia with or without relevant pretreatments.
Simulated ischemia
We have developed a model of simulated ischemia that has previously been described. 23 Chronic ischemia is difficult to replicate in vitro; however, the aim of the simulated ischemia model was to create an in vitro environment that matched the parameters of chronic ischemia, such as PO2, CO2, pH, apoptosis, and lactate dehydrogenase release, which have been demonstrated in the skeletal muscle of patients with CLI. Briefly, 24 hours following culture media change, myotubes (in open dishes) were exposed to 20% CO2 + 80% N2 (British Oxygen, Luton, UK) in hypoxic chambers (Modular Incubator Chamber, MIC-101; Billups-Rothenberg, Del Mar, CA, USA). The chamber was flushed with gas at 10 L/min for 20 min and then sealed. The apparatus was placed in an incubator at 37°C for 8, 24, and 72 h. Control myotubes were maintained under normoxic conditions (21% O2 + 5% CO2) for 8 h. The oxygen tension of the ischemic media was 6.8 kPa compared to 16.2 kPa in control media. The media was collected and centrifuged at 1600 g for 5 min at 4°C and stored at −20°C. The myotube monolayer was harvested on ice, washed with cold PBS and, following this, 75 μL RIPA buffer 1 × solution (150 mM NaCl, 1.0% IGEPAL® CA-630, 0.5% sodium deoxycholate, 0.1% SDS, and 50 mM Tris, pH 8.0; product number R0278; Sigma Aldrich, St Louis, MO, USA), and 75 μL of Laemmli buffer was added to each well. The monolayer was scrapped with a cell scrapper and collected into a 1.5 mL Eppendorf tube using a syringe with a 23 G needle. A total of 5 μL (2–5%) of 2-mercaptoethanol was added to the cell extract, which was then heated at 95°C for 5 min. Extracts were then centrifuged at 1600 g for 5 min at 4°C. The supernatant was collected and stored at −80°C for western blot analysis.
Pretreatment studies
To investigate the role of TLR2 and TLR6, and the downstream signaling pathways involved in ischemia-induced apoptosis and IL-6 induction, the following agonist and neutralising antibodies/inhibitors were used: TLR2/6 agonist PAM2CSK4 (10 μg/mL, EMC-MicroCollections; Tuebingen, Germany) was incubated with myotubes for 8 h; anti-TLR2 antibody (50 μg/mL, T2.5; eBioscience, San Diego, CA, USA) and anti-TLR6 antibody (1.25 μg/mL, pab-hstlr6; Invivogen, San Diego, CA, USA) were added to the myotubes 30 min prior to incubating in ischemia or added with PAM2CSK4; MyD88 inhibitor (100 μg/mL, tlrl-pimyd; Invivogen) was added 24 h prior to incubating in ischemia; TRIF inhibitor (20 μM, tlrl-pitrif; Invivogen) was added 6 h prior to incubating in ischemia; and NF-κB inhibitor (10 μM, Celastrol ant-cls; Invivogen) was added just before incubating in ischemia (all based on dose-ranging experiments; data not shown).
Immunocytochemistry
TLR2 and TLR6 co-localization
OCT-embedded gastrocnemius muscle was cut into 6 μm sections and mounted for double-immunofluorescence staining. Sections were fixed with ice-cold acetone and blocked with 10% chicken serum. Following this, sections were incubated in rabbit anti-human TLR2 antibody (1:50, sc-10739; Santa Cruz Biotechnology, Santa Cruz, CA, USA) and goat anti-human TLR6 antibody (1:50, sc-5657; Santa Cruz Biotechnology). After incubation with primary antibody or its IgG isotype control for 1 h at room temperature, slides were washed with PBS and treated with Texas Red® chicken anti-rabbit (1:200; Invitrogen) and FITC-chicken anti-goat (1:200; Invitrogen) secondary antibodies. The slides were washed with phosphate buffered saline (PBS) and coverslips were mounted using VECTASHIELD® mounting medium containing DAPI. The images were viewed using a fluorescence microscope (Axioscop 2; Carl Zeiss Micro-imaging, Germany).
Apoptosis
Cleaved caspase-3 expression was used to assess apoptosis. Chamber slides were removed from the hypoxic chamber and the myotube monolayer was fixed in 4% formaldehyde for 5 min. Serum block was performed with 10% goat serum and cells were incubated with cleaved caspase-3 (Asp175) antibody (1:50; Cell Signaling Technology, Danvers, MA, USA) or its IgG isotype control for 1 h at room temperature. Slides were then treated with Texas Red-X goat anti-mouse IgG antibody (Invitrogen) in 1:1000 dilution for 60 min in the dark at room temperature. The slides were washed with PBS and coverslips were mounted using VECTASHIELD mounting medium containing DAPI. The images were viewed using a fluorescence microscope.
Western blot analyses
Equal amounts of denatured protein were loaded per lane (24 μg) and were separated on 12% Tris-glycine gel followed by transfer onto a nitrocellulose membrane (Hybond-C extra; GE Healthcare Life Sciences, Buckinghamshire, UK). The membranes were blocked with 5% milk in PBS/0.1% Tween 20 and incubated overnight at 4°C with the following antibodies: TLR2 (1:500, sc-10739; Santa Cruz), TLR6 (1:500, sc-5661; Santa Cruz), TLR6 (1:500, ab-71429; Abcam, Cambridge, UK), NF-κB (1:1000, C22B4; Cell Signaling), Phospho-NF-κB (1:2000, 93H1; Cell Signaling); IκBα (1:1000; Cell Signaling); Phospho-IκBα (1:1000; Cell Signaling); cleaved caspase-3 (1:1000, 9661; Cell Signaling); BAX (1:500, sc-526; Santa Cruz), BCL2 (1:500, sc-783; Santa Cruz), HMGB-1 (1:1000, ab18256; Abcam), and β-tubulin-loading control (1:10,000, ab4074; Abcam). The membranes were then washed and incubated with horseradish peroxidase-linked corresponding secondary antibody at room temperature. The membrane was developed using chemiluminescent substrate (Amersham ECL Plus™ Western Blotting Detection Reagent; GE Healthcare Life Sciences) and the blots were developed against photographic film (Amersham Hyperfilm ECL, GE Healthcare Life Sciences). Band intensities were determined by densitometry using the Biospectrum® AC imaging system (UVP, Upland, CA, USA).
Immunoprecipitation
Ischemic and control C2C12 myotube monolayers were lysed with lysis buffer (1% SDS, 1.0 mM sodium-orthovandate, 10 mM Tris pH 7.4). The lysate was than boiled for 5 min, sonicated and centrifuged. Then 100 μL of lysate was incubated at 4°C for 1 h with 2 μg anti-TLR2 (sc-10739) or anti-TLR6 (sc-5661), 400 μL H2O, and 2X immunoprecipitation buffer (2% Triton X-100, 300 mM NaCl, 20 mM Tris pH 7.4, 2 mM EDTA, 2 mM EGTA pH 8.0, 0.4 mM sodium orthovanadate, 0.4 mM PMSF, 1% NP-40). Subsequently, the lysate was incubated with 50 μL protein G-Sepharose (P3296; Sigma-Aldrich, Dorset, UK) for 30 min at 4°C with agitation. Bound proteins were than washed twice with 1X immunoprecipitation buffer and the pellet resuspended in 45 μL 2X laemlli buffer. Immune complexes were separated by Tris-glycine, transferred to nitrocellulose, and immunoblotted with TLR2 or TLR6 antibodies. Following optimisation of the antibody and the technique for the immunoprecipitation studies, we did not include IgG control for further experiments. Proteins were visualised by using chemiluminescence followed by exposure to photographic film.
IL-6 assay
IL-6 protein concentrations in the culture media were determined using an ELISA kit (Mouse IL-6 ELISA Ready-SET-Go!®; eBioscience, San Diego, CA, USA) following the manufacturer’s protocol. Samples and known standards were assayed in triplicate. The lower detection limit was 4 pg/mL.
Statistical analysis
Data were analysed with GraphPad Prism software (version 5, GraphPad, San Diego, CA, USA). Parametric data were compared using a two-tailed Student’s t-test and analysis of variance (ANOVA)/Tukey’s test; values represent mean ± SE. Non-parametric data were described using median ± range and were compared using a two-tailed Mann–Whitney U test. A p-value of < 0.05 was considered statistically significant.
Informed consent statement
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2000. Written informed consent was obtained from patients with prior approval from the Royal Free & Medical School Research Ethics Committee (REC ref: 29-2000).
No animal studies were carried out by the authors for this article.
Results
TLR2 and TLR6 protein expression is upregulated in critically ischemic muscle and in C2C12 myotubes exposed to simulated ischemia
TLR1–7 have been shown to be expressed in skeletal muscle 9 and a number of studies have demonstrated that their expression is upregulated following ischemia in various organ systems. However, previous studies have not looked at the effect of skeletal muscle ischemia on TLR expression. We found a significant upregulation of TLR2 (p = 0.004) and TLR6 (p = 0.002) protein expression in muscle from patients with CLI (Figure 1A and 1C). During our optimisation of the in vitro ischemia model, we investigated different durations of hypoxia, thus exposing the myotubes to 8 h, 24 h, and 72 h. We found that 8 h was the optimum duration for the myotubes to be exposed to ischemia in terms of ischemic parameters such as the pH, PO2, CO2, and lactate. Longer periods of exposure to hypoxia began to effect the viability of the myotubes. We observed that 8 h and 24 h of ischemia did not have a significant effect on TLR2 and TLR6 expression (data not shown). However, myotubes exposed to 72 h of ischemia showed a significant upregulation of TLR2 (p = 0.006) and TLR6 (p = 0.0014) protein expression (data not shown).
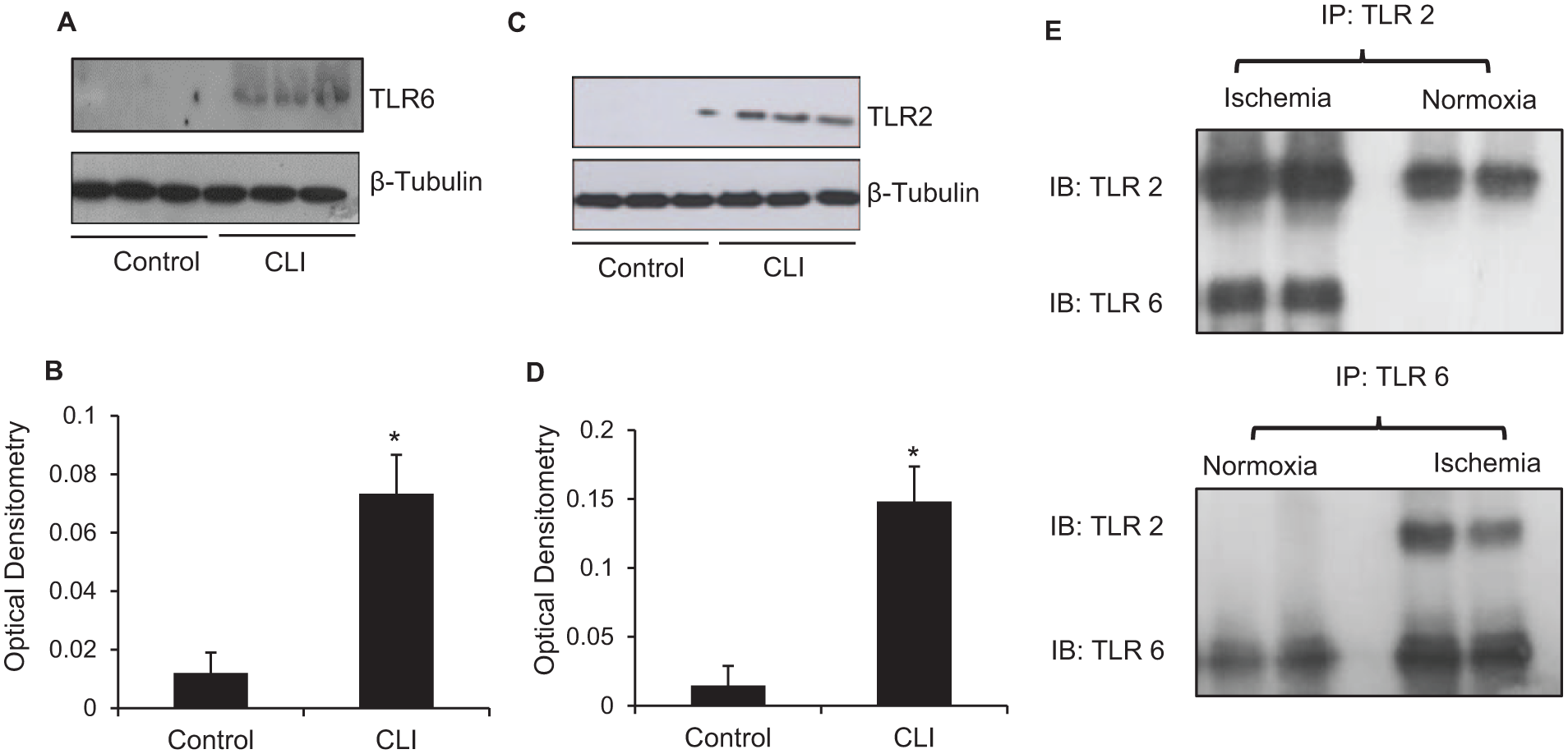
TLR2 and TLR6 expression in ischemic muscle. (A, C) Representative TLR2 and six western blots of control and CLI patient muscle samples. Blot was reprobed for β-tubulin expression as control for protein loading. (B, D) Densitometric quantification of TLR2 and six levels in CLI muscle. (E) TLR2 heterodimerises with TLR6 following ischemia.
TLR2 and TLR6 co-localize and co-immunoprecipitate in muscle following ischemia with subsequent activation of the TLR signaling pathway
TLR2 heterodimerises with TLR1 or TLR6 depending on the ligand that is presented. We found increased TLR2 and TLR6 co-localization in ischemic human muscle (data not shown) as well as co-immunoprecipitation of TLR2 and TLR6 following ischemia (Figure 1E). This would suggest that ischemia leads to TLR2 and TLR6 heterodimerisation and activation of the signaling pathway. In its inactive form, NF-κB is bound to the inhibitory complex IκBα. Following TLR pathway activation, IκBα is phosphorylated and NF-κB is released. Further, optimal NF-κB activation occurs following phosphorylation of the p65 subunit of NF-κB. Considering these principles, we assessed protein expression of IκB and NF-κB (p65) as well as their phosphorylated forms in control and ischemic C2C12 myotubes. Protein expression of both IκB (p = 0.0028; Figure 2A) and NF-κB (p = 0.003; Figure 2C) was reduced in ischemic myotubes. Conversely, there was significantly increased expression of the phosphorylated form of IκBα (p < 0.0001; Figure 2A) and NF-κB (p = 0.01; Figure 2C) in ischemic but not control myotubes. This indicated that simulated ischemia not only leads to TLR2 and TLR6 heterodimerisation, but also activation of the TLR signaling pathway and subsequent NF-κB activation by its phosphorylation.
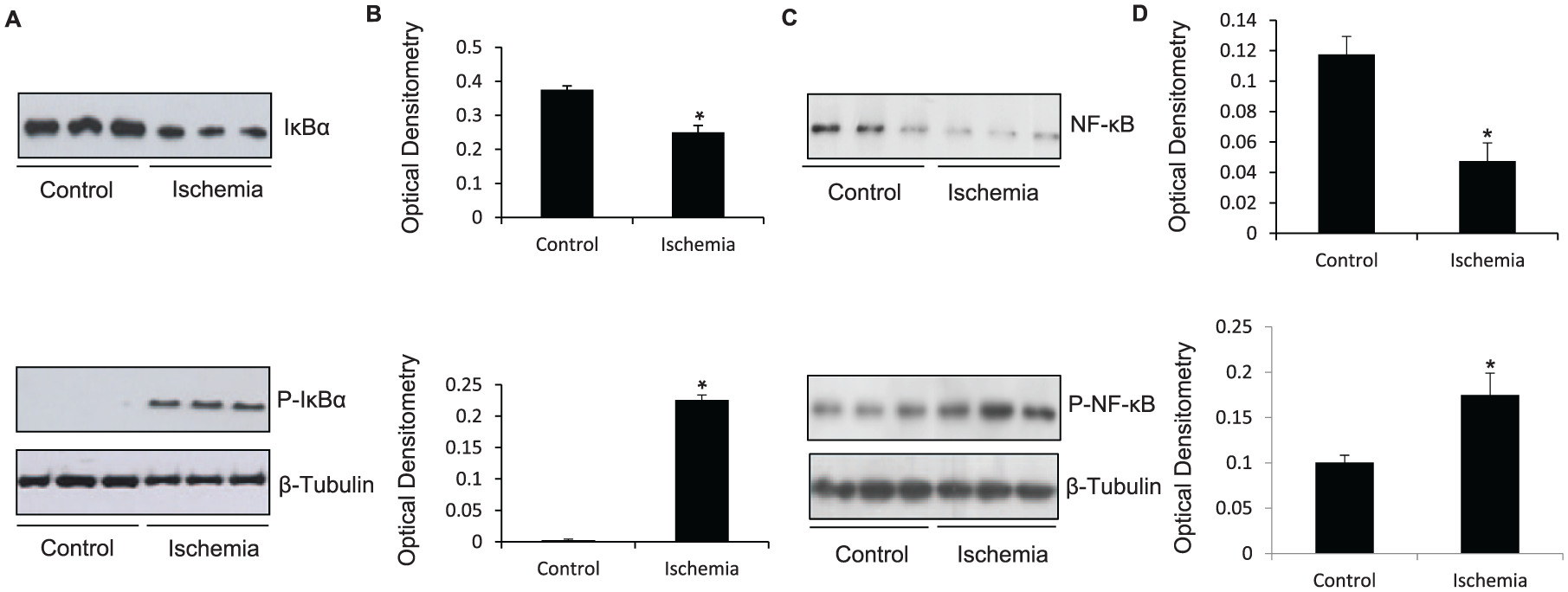
TLR2 and TLR6 downstream signaling pathway in ischemia. (A) Representative western blot showing downregulation of IκBα and upregulation of P-IκBα in C2C12 myotubes exposed to 8 h of simulated ischemia compared to control myotubes. Protein loading was consistent, as shown by β-tubulin levels. (B) Densitometric analyses of western blots. (C) Representative western blot showing downregulation of NF-κB and upregulation of P-NF-κB in C2C12 myotubes exposed to 8 h of simulated ischemia compared to control myotubes. Protein loading was consistent, as shown by β-tubulin levels. (D) Densitometric analyses of western blots.
TLR2 and TLR6 blockade reduces ischemia-induced apoptosis
Increased apoptosis has been demonstrated in skeletal muscle of patients with PAD. 1 We therefore evaluated the effect of ischemia on myotube apoptosis. We found that ischemia increased apoptosis, as shown by increased B-cell lymphoma 2 (BCL2)-associated X protein (BAX) (p = 0.005; Figure 3B) and decreased BCL2 (p = 0.002; Figure 3B) expression. This finding was confirmed by increased cleaved caspase-3 expression on western blot (Figure 3D) and immunofluorescence (Figure 3A). Further, neutralising anti-TLR2 (p < 0.01; Figure 3D) and anti-TLR6 (p < 0.01; Figure 3D) antibodies reduced ischemia-induced apoptosis, as represented by reduced cleaved caspase-3 protein expression on western blot. These findings suggest that ischemia-induced apoptosis is mediated via TLR2 and TLR6 activation in skeletal muscle.
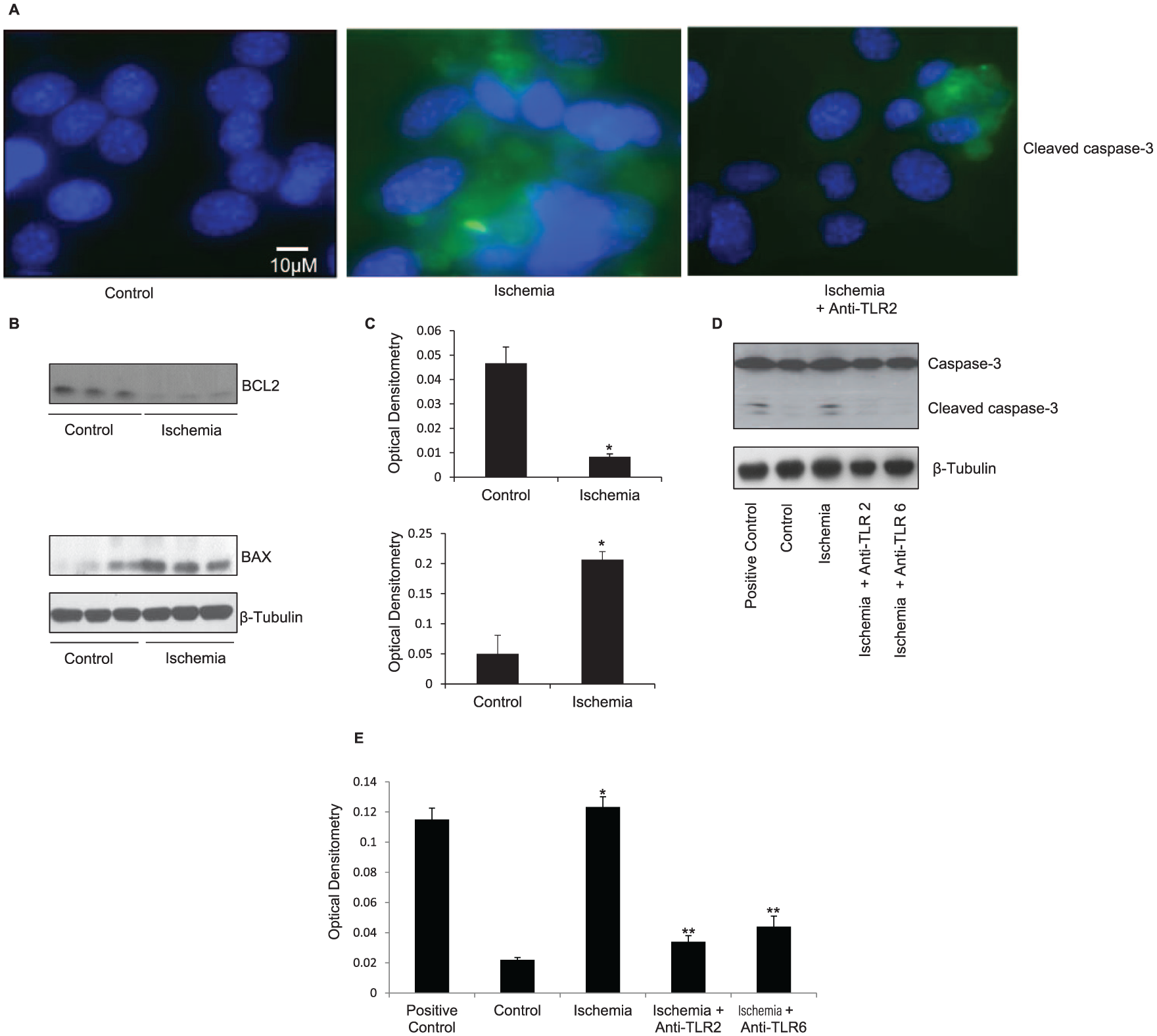
TLR2 and TLR6 blockade reduced ischemia-induced apoptosis. (A) Immunofluorescence staining for DAPI (blue) and cleaved caspase-3 (green) was performed on control and ischemic myotubes, as well as ischemic myotubes incubated with neutralising anti-TLR2 antibody. There is increased cleaved caspase-3 staining in the ischemic myotubes and significantly reduced cleaved caspase-3 staining in the ischemic myotubes with anti-TLR2 antibody. (B) Representative western blot showing downregulation of BCL2 and upregulation of BAX in C2C12 myotubes exposed to 72 h of simulated ischemia compared to control myotubes. Protein loading was consistent, as shown by β-tubulin levels. (C) Densitometric analyses of western blot. (D) Representative western blot showing increased cleaved caspase-3 expression following 8 h of simulated ischemia. Protein loading was consistent, as shown by β-tubulin levels. (E) Densitometric analyses of western blots.
Ischemia-induces skeletal muscle inflammation via TLR2 and TLR6
Patients with PAD have been reported to have high levels of circulating cytokines such as TNF-α and IL-6, 24 and ischemia itself is known to trigger the release of inflammatory cytokines in a variety of organs. We found that IL-6 release was significantly upregulated in the media of the ischemic myotubes (p < 0.01; Figure 4A). In addition, the TLR2 and TLR6 agonist PAM2CSK4 also increased IL-6 (p < 0.01; Figure 4A) release in the absence of an ischemic stimuli. Further, neutralising anti-TLR2 (p < 0.01) and TLR6 (p < 0.01) antibodies both reduced the ischemia-induced IL-6 release, indicating that the IL-6 release was mediated via the activation of these two receptors (Figure 4A).
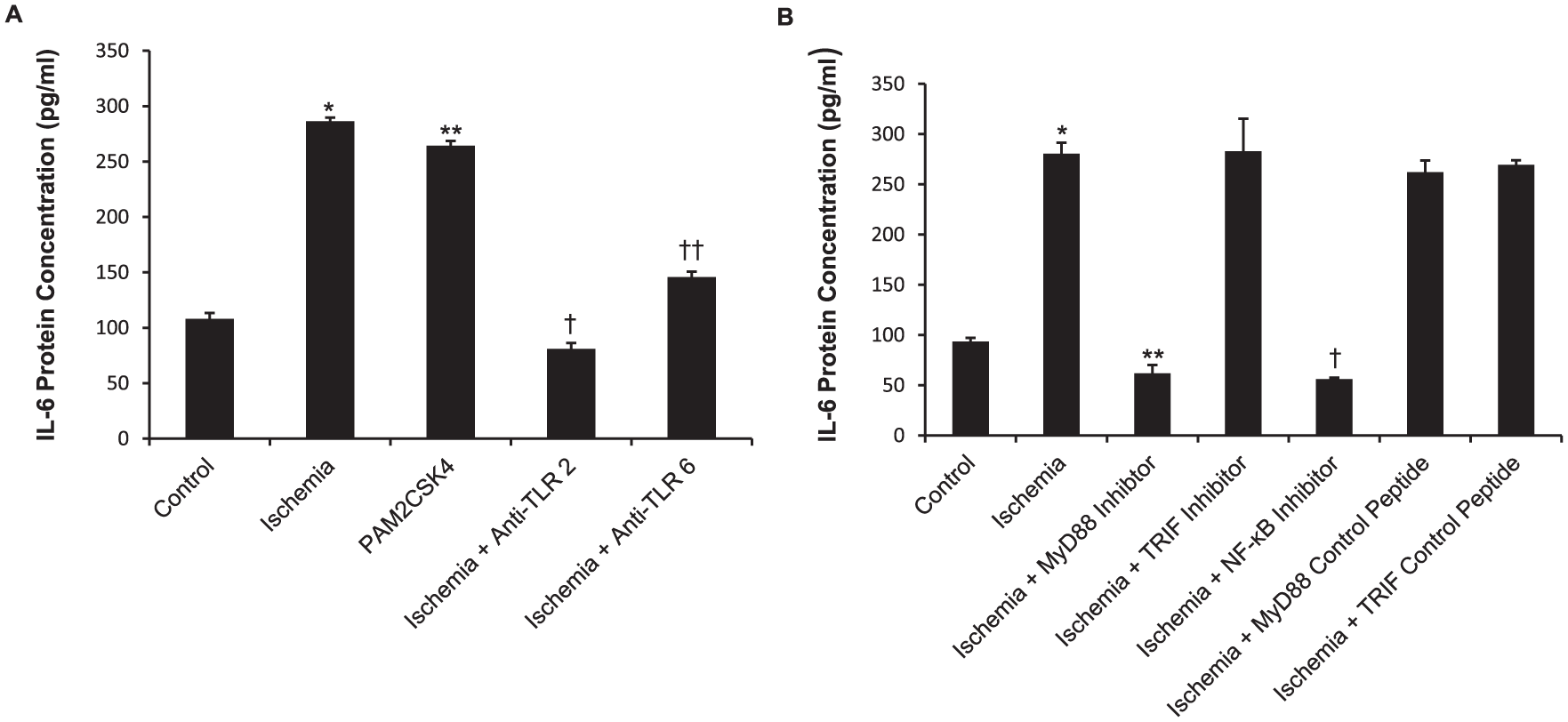
Simulated ischemia increases IL-6 secretion via TLR2 and TLR6. (A) IL-6 generation in C2C12 myotube supernatant was measured by ELISA. Myotubes were exposed to simulated ischemia for 8 h with or without anti-TLR2 or anti-TLR6 antibodies. (B) IL-6 generation in C2C12 myotube supernatant was measured by ELISA. Myotubes were pretreated with specific inhibitors or control peptides and exposed to simulated ischemia for 8 h.
Ischemia-induced IL-6 generation and apoptosis is mediated via MyD88 signaling pathway and NF-κB activation
TLR signal transduction is mediated via the MyD88-dependent and MyD88-independent pathways and this leads to the activation of a number of transcription factors such as NF-κB and AP-1. We sought to determine whether both pathways are utilised in ischemia-induced apoptosis and IL-6 release and whether IL-6 generation is a result of NF-κB activation. C2C12 myotubes were pretreated with a MyD88, TRIF or NF-κB inhibitor prior to exposure to 8 h of simulated ischemia. The C2C12 myotubes pretreated with the MyD88 (p < 0.01) and NF-κB (p < 0.01) inhibitors showed reduced IL-6 generation, whilst TRIF inhibition as well as the MyD88 and TRIF control peptides did not reduce ischemia-induced IL-6 release (Figure 4B). Further, MyD88 inhibition but not TRIF inhibition reduced ischemia-induced apoptosis, as shown by the reduced cleaved caspase-3 expression (Figure 5A). MyD88 inhibition disrupted ischemia-induced NF-κB phosphorylation and hence activation; however, TRIF inhibition did not abrogate ischemia-induced NF-κB activation (Figure 5C).
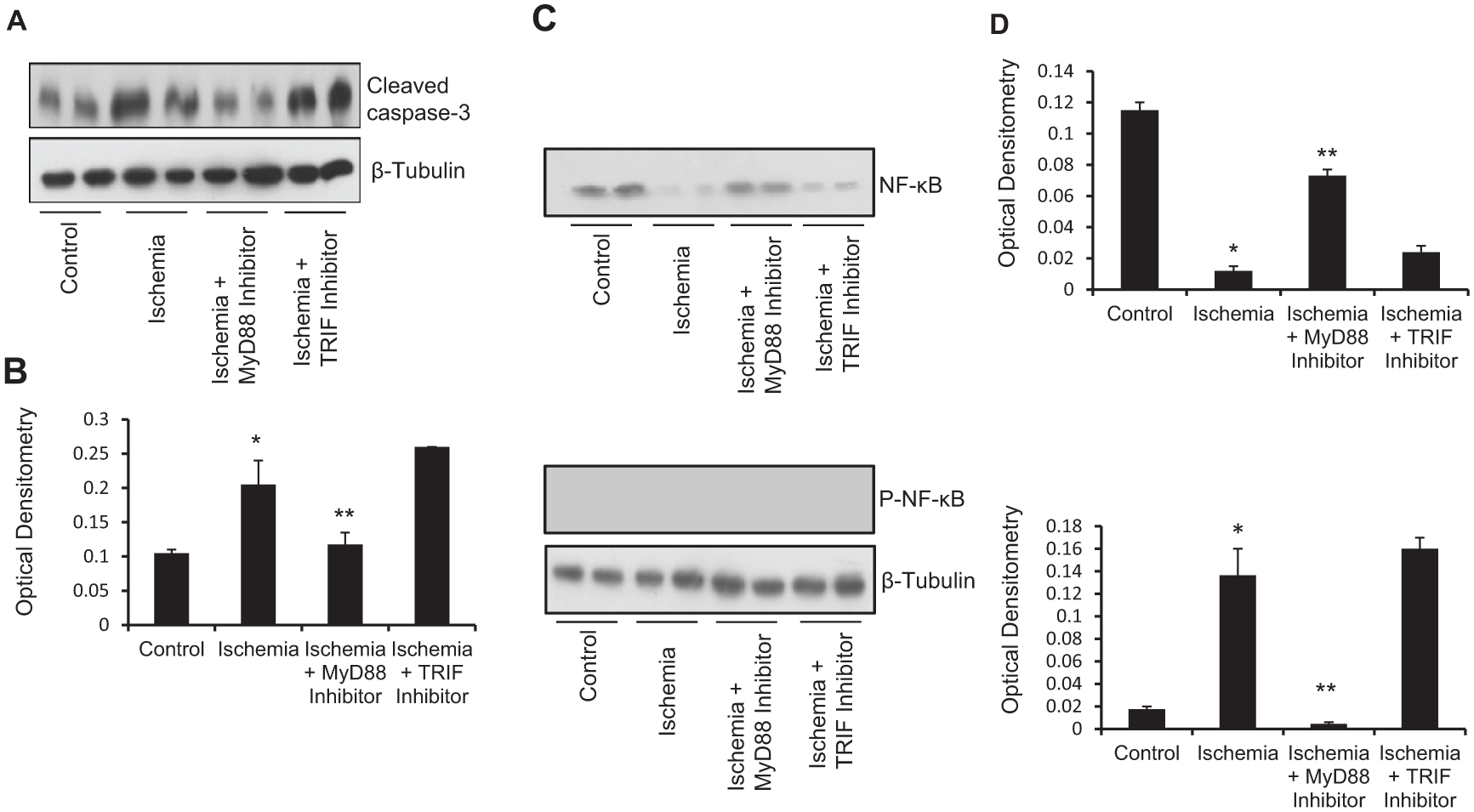
Ischemia-induced apoptosis and NF-κB activation is mediated via the MyD88 signaling pathway. (A) Representative western blot showing decreased cleaved caspase-3 expression. MyD88 inhibition significantly reduced ischemia-induced cleaved caspase-3 expression. Protein loading was consistent, as shown by β-tubulin levels. (B) Densitometric analyses of western blots. (C) Representative western blot showing NF-κB and P-NF-κB expression in C2C12 myotubes exposed to 8 h of simulated ischemia. Protein loading was consistent, as shown by β-tubulin levels. (D) Densitometric analyses of western blots.
Potential endogenous ligands
Among the endogenous ligands or damaged-associated molecular patterns (DAMPs) that activate TLRs, HMGB-1 and the HSPs have been implicated in activating TLRs following an ischemic insult. We therefore determined the expression of HMGB-1 and HSPs 60 25 and 70 26 in ischemic C2C12 myotubes. We have found that following an ischemic insult there was an increase in the expression of HMGB-1 (p = 0.007; Figure 6A), HSP 60 (p = 0.0003; Figure 6C), and HSP 70 (p < 0.0001; Figure 6C)
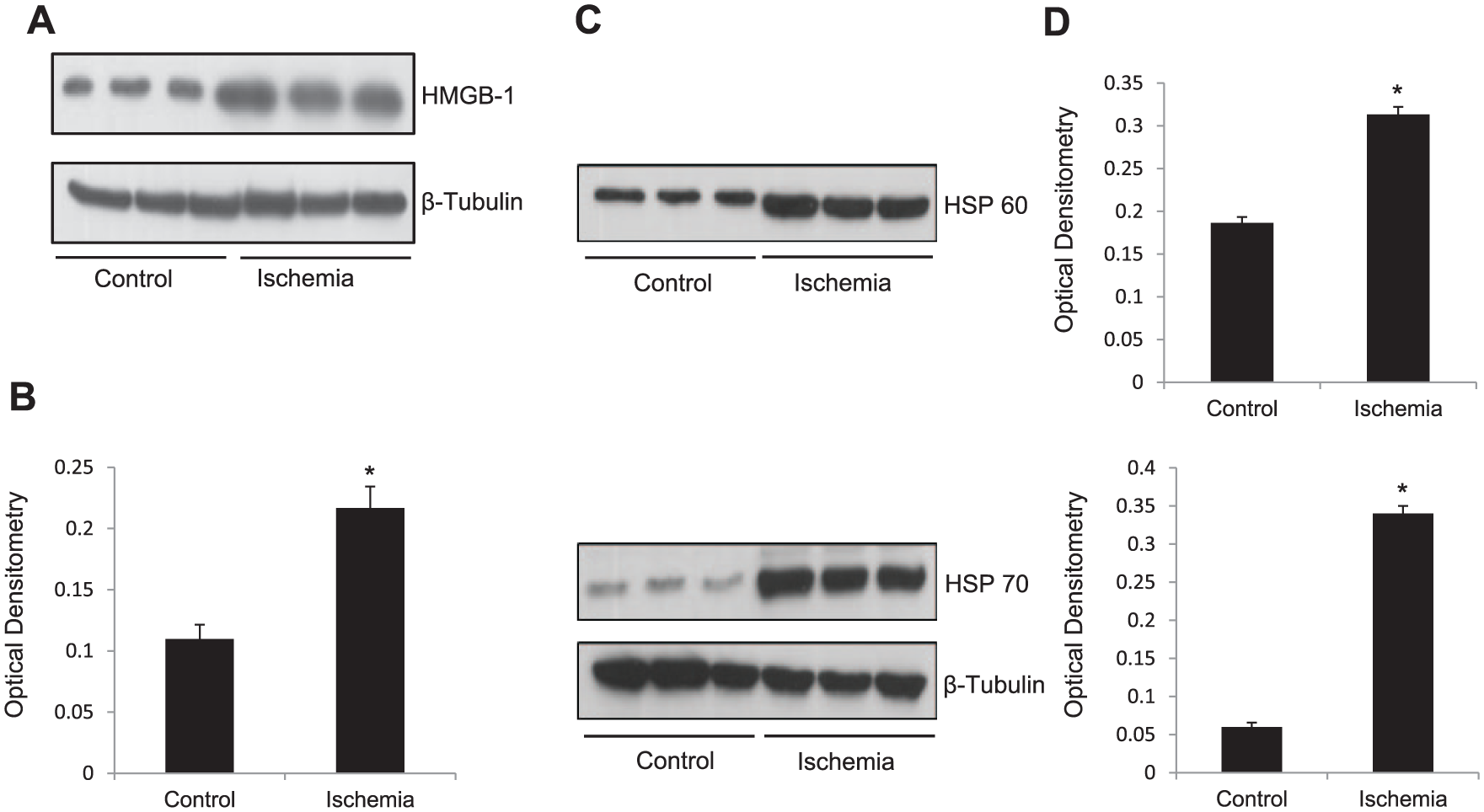
Simulated ischemia increased the release of endogenous ligands HMGB-1, HSP 60, and HSP 70. (A) Representative western blot showing upregulation of HMGB-1 in C2C12 myotubes exposed to 8 h of simulated ischemia compared to control myotubes. Protein loading was consistent, as shown by β-tubulin levels. (B) Densitometric analyses of western blots. (C) Representative western blot showing upregulation of HSPs 60 and 70 in C2C12 myotubes exposed to 8 h of simulated ischemia compared to control myotubes. Protein loading was consistent, as shown by β-tubulin levels. (D) Densitometric analyses of western blots.
Discussion
The management of CLI is predominantly aimed at improving blood flow or in severe cases major amputation is required to prevent progression of gangrene. However, despite successful improvement in blood flow to the ischemic leg, many patients still complain of no improvement of symptoms in terms of pain, exercise tolerance, and quality of life and therefore deem the surgery to be unsuccessful. The exact pathophysiology as to why these patients have ongoing muscle dysfunction despite the restoration of blood flow is unclear. However, emerging evidence suggests that this may be due to ischemia-induced skeletal muscle damage and it is thought that inflammation plays a central role in this process. 27 In support of the theory that inflammation plays an important role in skeletal muscle damage in CLI, it has been shown that raised levels of circulating pro-inflammatory cytokines such as TNFα, IL-6 and chemokines are found in patients with PAD. 24
The discovery of TLRs and their role in immunity and inflammation has led to significant advances in terms of understanding the pathophysiology of ischemia and ischemia/reperfusion-mediated organ injury. In particular, TLR2 and TLR4 have been strongly implicated in myocardial injury following sustained ischemia, and TLR2 28 and TLR4 29 knockout mice have been shown to have reduced myocardial infarct size. These emerging studies along with the fact TLRs are also expressed on skeletal muscle, provide a potential understanding of how ischemia in CLI patients causes skeletal muscle damage.
Preliminary studies using focused RT-PCR microarray studies carried out in our lab demonstrated upregulation of gene expression of cell surface TLRs 1, 2, 4, and 6, as well as TLR-related cofactors MyD88 and CD14, and downstream targets IL-6, IL-8, and IRAK1 in muscle from CLI patients. TLR2 upregulation was particularly marked; further, TLR2 and TLR6 heterodimerisation has been described. 30 Therefore, we decided to investigate these two TLRs further. In the present study, we have not only shown that TLR2 and TLR6 are expressed on human skeletal muscle but that their expression is increased in ischemic skeletal muscle. We observed from immunofluorescence staining that TLRs are expressed on endothelium (CD31), neutrophil (CD43), and macrophages (CD68) within muscle tissue. Given this widespread expression, as well as known expression of TLRs on muscle cells, we decided to focus here on the effect of ischemia on muscle tissue TLR expression, where the TLR activation on individual cell types are likely to exert an autocrine/paracrine effect on the tissue as a whole. In order to understand the significance of this finding we have utilised an in vitro model of simulated ischemia previously developed by our group. Significantly, TLR2 and TLR6 expression was upregulated in the ischemic C2C12 myotubes. These findings corroborate with other studies which have shown that ischemia upregulates TLRs in various tissues both in vivo and in vitro. Our experiments have demonstrated that TLR2 and TLR6 also heterodimerise under ischemic conditions, suggesting ligand binding and activation of the downstream signaling pathway. Indeed, we found that IκBα was phosphorylated following an ischemic insult, indicating activation of NF-κB, which is central to the translocation of NF-κB into the nucleus and the subsequent induction of pro-inflammatory cytokines.
Several studies have shown that following ischemia, cellular death results due to apoptosis, and increased apoptosis of skeletal muscle has also been demonstrated in patients with PAD. Our data supports that an ischemic insult upregulates cellular apoptosis in muscle. We have also shown that ischemia-induced apoptosis in skeletal muscle is reduced by neutralising anti-TLR2 and TLR6 antibodies, indicating that ischemia-induced apoptosis is mediated via TLR2 and TLR6. The mechanism for this is still poorly understood but studies have shown that TLR2 may promote apoptotic cell death via several pathways, such as activation of the JNK-AP-1 31 pathway or via a pathway involving Fas-associated death domain protein (FADD) and caspase 8. 14
IL-6 is a potent inflammatory cytokine that plays a significant role in the pathogenesis of cell damage following ischemia in various tissues. We have established that ischemia results in an increase in IL-6 release by myotubes whilst inhibiting TLR2 and TLR6-reduced IL-6 production. This would suggest that TLR2 and TLR6 are important in the ischemia-induced upregulation of IL-6 and any subsequent muscle damage propagated by this inflammatory cytokine.
We further sought to elucidate which TLR signaling pathway is implicated in these potentially pathological processes. We found that ischemia-induced NF-κB phosphorylation, apoptosis, and IL-6 production in skeletal muscle is mediated via the MyD88-dependent and not the MyD88-independent (TRIF-dependant) pathway. The MyD88 pathway may therefore be a potential therapeutic target in reducing ischemic muscle damage. In addition, by targeting a specific pathway, potential side effects of any medical therapy would be kept to a minimum.
Several TLR2 and TLR6 endogenous ligands have been identified that are released in response to stress. In particular, HMGB-1, HSP 60, and HSP 70 have already been implicated in the pathogenesis of renal, liver, and myocardial damage following ischemia. We have shown in our previous studies that there is upregulation in the expression of HMGB-1, HSP 60, and HSP 70 following ischemia; further, HMGB-1 and HSP 60 have been shown to co-localize with TLR2 and TLR4.30,32 Therefore, one can speculate that these endogenous ligands may be activating TLR2 and TLR6 and propagating the skeletal muscle damage in patients with limb ischemia. However, we have yet to identify whether these potential endogenous ligands specifically activate TLR2 and TLR6 following skeletal muscle ischemia or whether there is an alternative mechanism for their activation. Recombinant HMGB-1 has been shown to worsen myocardial injury following ischemia/reperfusion, whereas treatment with the HMGB-1 antagonist HMGB-1 box A reduced infarct size. 11 Thus, it would be interesting to see whether antagonising HMGB-1, HSP 60 or HSP 70 may reduce skeletal muscle damage following an ischemic insult.
Limitations
This study has several limitations. Analyses of our muscle biopsies from patients provided only observational evidence for the involvement of TLR2 and TLR6 in ischemic skeletal muscle. Further investigation was carried out in an in vitro model where C2C12 myotubes were used. This is an established murine cell line, which may not fully reflect the situation in human cells. In vitro models are also not representative of the complex in vivo conditions that occur. However, using a combination of patient tissue analyses and in vitro experiments has allowed us to start elucidating potential pathways for more detailed investigation. As previously mentioned, other TLRs have been shown to play a role in ischemia-induced damage. 16 In particular, TLR2 has also been shown to heterodimerise with TLR1 under certain conditions. It would not be surprising that the other TLRs may also be playing a role in skeletal muscle injury as the functional 11 TLRs mediate their signaling via the same pathways. Therefore, whilst we have shown that TLR2 and TLR6 play a role in ischemia-induced skeletal muscle damage, future studies focusing on the other TLRs, in particular TLR4, would be beneficial.
Conclusion
In conclusion, we have shown that TLR2 and TLR6 are upregulated in skeletal muscle following ischemia. The receptors heterodimerise, leading to activation of the MyD88 signaling pathway with subsequent activation of NF-κB. This results in upregulation of the inflammatory cytokine IL-6 as well as an increase in muscle apoptosis. It is most likely that this results in an environment whereby the inflammatory milieu leads to a myopathy and thus limits the therapeutic effect of revascularisation. Our study has identified potential therapeutic targets such as the MyD88 signaling pathway, which may be blocked in order to prevent or reduce muscle damage. It is possible that medical therapy with the aim of reducing skeletal muscle damage in conjunction with improvement in blood flow may provide symptomatic relief and improve functional outcomes.
Supplemental Material
10.1177_1358863X19843180_Supplementary_Figure1 – Supplemental material for Toll-like receptors 2 and 6 mediate apoptosis and inflammation in ischemic skeletal myotubes
Supplemental material, 10.1177_1358863X19843180_Supplementary_Figure1 for Toll-like receptors 2 and 6 mediate apoptosis and inflammation in ischemic skeletal myotubes by Hemanshu Patel, Cissy Yong, Ali Navi, Sidney G Shaw, Xu Shiwen, David Abraham, Daryll M Baker and Janice CS Tsui in Vascular Medicine
Footnotes
Acknowledgements
We thank the operating theatre staff of The Heart Hospital and the Royal Free Hospital for helping with tissue collection.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: H Patel was supported by Circulation Foundation research fellowship. J Tsui and S Shaw were supported by a Royal Society International Joint Project grant.
Supplementary material
The supplementary material is available online with the article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.