
Abstract
The recruitment and homing of circulating bone marrow-derived cells include endothelial progenitor cells (EPCs) that are critical to neovascularization and tissue regeneration of various vascular pathologies. We report here that conditional inactivation of hypoxia-inducible factor’s (HIF) transcriptional activity in the endothelium of adult mice (ArntΔiEC mice) results in a disturbance of infiltrating cells, a hallmark of neoangiogenesis, during the early phases of wound healing. Cutaneous biopsy punches show distinct migration of CD31+ cells into wounds of control mice by 36 hours. However, a significant decline in numbers of infiltrating cells with immature vascular markers, as well as decreased transcript levels of genes associated with their expression and recruitment, were identified in wounds of ArntΔiEC mice. Matrigel plug assays further confirmed neoangiogenic deficiencies alongside a reduction in numbers of proangiogenic progenitor cells from bone marrow and peripheral blood samples of recombinant vascular endothelial growth factor-treated ArntΔiEC mice. In addition to HIF’s autocrine requirements in endothelial cells, our data implicate that extrinsic microenvironmental cues provided by endothelial HIF are pivotal for early migration of proangiogenic cells, including those involved in wound healing.
Keywords
Background
Neoangiogenesis or vasculogenesis, originally thought to be exclusive to embryogenesis, involves the de novo production of endothelial progenitor cells known as angioblasts and their formation into a vascular plexus that progressively matures by various approaches of remodeling during the angiogenic phase.1,2 However, in more recent years, vasculogenesis has been recognized as an important process to adult vascular responses. 3 Postnatally, angiogenesis engages the activation of quiescent endothelial cells (ECs) found in pre-existing vessels, while neoangiogenesis requires the recruitment of bone marrow (BM)-derived proangiogenic progenitor cells (BMDPPCs) first described as circulating endothelial progenitor cells (EPCs).4,5 While the identification and characterization of EPCs remain controversial, evidence argues that the majority of BMDPPCs primarily arise from a stem cell pool within the BM homing to injured tissues in response to hypoxia and chemokines.6–9
Hypoxia-inducible factor (HIF) is a master heterodimeric transcriptional regulator that responds to changes in oxygen (O2) tension. While either HIF-1α or -2α subunits are constitutively expressed, in the presence of O2 they are subjected to prolyl hydroxylation, ubiquitination, and proteasomal degradation. 10 In response to hypoxia, hydroxylation is inhibited and the α-subunits heterodimerize with ARNT (aryl hydrocarbon nuclear translocator, HIF-1β), activating multiple genes containing hypoxic response elements (HREs). 11 Previous work from multiple laboratories showed that loss of either HIF α-subunit in the endothelium leads to vascular defects.12–14 While a hypoxic environment in pathological tissues may mediate the recruitment and homing of BMDPPC, exactly how ECs may contribute to neoangiogenic responses has not been effectively addressed.15–19
In this study, we fully inactivated HIF’s transcriptional activity in adult VE-cadherin+ ECs and identified a critical role for HIF in the progress of vasculogenic responses. More specifically, we show herein the absence of endothelial-ARNT results in reduced vascular BMDPPC accumulation and neovascularization that ultimately delay dermal wound healing.
Methods
Generation of ArntΔiEC mice
All animal experiments were performed in accordance with approved animal protocols from the Case Western Reserve University Animal Care and Use Committee and National Institutes of Health guidelines. Arnt-conditional (obtained from F. Gonzalez) 20 and inducible-VecadCreERT2 (obtained from L. Iruela-Arispe) 21 mice were crossed to obtain ArntloxP/loxP::VecadCreERT2 experimental mice. In all experiments, littermates from the same breeding pair were used as controls. Tamoxifen (Sigma) was dissolved in a sunflower oil/ethanol (10:1) mixture at 20 mg/ml. Tamoxifen 1 mg was injected daily intraperitoneally into 8-week-old ArntloxP/loxP::VecadCreERT2 mice and control littermates (either ArntloxP/+::VecadCreERT2 or ArntloxP/loxP) for 10 consecutive days and Arnt-deletion was detected by PCR. 22
Irradiation and bone marrow transplantation
ArntloxP/loxP and ArntΔiEC recipient mice (11–14 weeks old) were sublethally irradiated, and bone marrow donor cells from UBC-GFP mice (Jackson Laboratory) were transplanted via tail vein injection.
Matrigel plug assay
Growth factor-reduced Matrigel (BD Biosciences) was combined in solution with recombinant vascular endothelial growth factor (VEGF) (200 ng/ml), fibroblast growth factor (FGF)-2 (1 μg/ml) and 60 U/ml heparin (Sigma). The gel solutions (500 μl each) were uniformly injected subcutaneously into the abdominal region of anesthetized mice. Matrigel plugs were harvested on day 7 and directly frozen in optimum cutting temperature (OCT) compound (Tissue-Tek) for immunohistochemistry. A hypoxic environment was detected by pimonidazole uptake using Hypoxyprobe™ Kits (HPI, Inc.). To aid visualization of neovessel organization and competency, 100-μl fluorescein isothiocyanate (FITC)-conjugated dextran (Mr 2×106; Sigma, 25 mg/ml) was administered intravenously via the right jugular vein 10 minutes before plug harvest.
Wound healing assay
The detailed procedure for wound-healing experiments was followed as previously described.23,24 Briefly, sex- and age-matched ArntloxP/loxP and ArntΔiEC mice were shaved and depilated. Mice were anesthetized and two full-thickness wounds were made with 6-mm skin biopsy punches; wounds were measured every 2–3 days until closure. Alternatively, wound tissues were harvested at days 1.5, 3, and 7 for RNA isolation and histology.
EPC mobilization
To further induce the mobilization of EPCs, a subset of sex- and age-matched ArntloxP/loxP and ArntΔiEC mice were pretreated daily with recombinant VEGF (Cell Signaling) in phosphate buffered saline (PBS; 100 mg/kg/day, in 100 µl) for 4 days prior to Matrigel injections. Matrigel plugs were harvested on day 7 and directly frozen in OCT compound (Tissue-Tek) for immuno-histochemistry. BM cells were also collected from these VEGF-treated ArntloxP/loxP and ArntΔiEC mice for further flow cytometry and colony-forming unit (CFU) analysis, and femur bones were processed for histology.
Hemogenic progenitor assays
Following 4 days of VEGF injections, animals were sacrificed and cells from BM and peripheral blood were plated on 6- or 48-well culture dishes (5×105 per well, respectively) coated with human fibronectin (Sigma) in endothelial basal medium (EBM) (Cell Systems) supplemented with endothelial growth medium SingleQuots and 10% fetal calf serum (FCS). After 4 days, non-adherent cells were removed, cultured for another 3 days, and then underwent cytochemical analysis. Individual EPC clusters were analyzed with standard staining (1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine-labeled acetylated LDL (DiLDL, 10 µg/mL) and FITC-labeled Ulex europaeus agglutinin I (lectin, 10 µg/mL; Sigma) for 1 hour and double-stained cells were quantified in three randomly selected fields per well. To confirm an endothelial phenotype of longer culturing of cells, a subset of wells was fixed with 2% paraformaldehyde for 10 minutes and double stained with CD31 and Alexa Fluor® 594-labeled Ulex europaeus agglutinin I (10 µg/mL) for 1 hour.
Mouse BM cells were obtained by flushing femoral and tibia bones with a 23-gauge needle containing cold 2% FCS/PBS and then red blood cells were lysed. Lin– BM cells were separated using a lineage cell depletion kit through magnetic columns (Miltenyi Biotec, Inc.) and a cocktail of biotinylated lineage-specific antibodies (CD5, CD45R (B220), CD11b, Gr-1 (Ly-6G/C), 7-4, and Ter-119). Mononuclear peripheral blood cells were isolated using Histopaque (Sigma). For HSC isolation and characterization, 1×105 BM cells were plated in mouse Methylcellulose Complete Media (R&D Systems). Colonies were counted and analyzed at day 10. EPC analysis was adapted from Masuda et al. 25 Briefly, 1×105 BMMNCs were plated in mouse Methylcellulose Base Media (R&D Systems) supplemented with 20% FBS, VEGF (50 ng/µl), basic FGF (bFGF) (25 ng/µl), epidermal growth factor (EGF) (5 ng/µl), insulin-like growth factor (IGF)-1 (50 ng/µl), stem cell factor (SCF) (50 ng/µl), interleukin (IL)-3 (15 ng/µl) and heparin (20 units/ml). EPC colonies were counted at day 10, replated and cultured on fibronectin-coated chamber slides in EC growth medium (EGM)-2 media (Lonza) to confirm EC potential by staining with CD31 and Flk-1.
Histology and immunohistochemistry
Wounds and Matrigel plugs were processed for standard frozen OCT sectioning. Frozen Matrigel plugs and wound samples were sectioned at 10-μm thickness followed by brief fixation in 4% paraformaldehyde (PFA). The fixed sections were reacted with primary antibodies and followed by the appropriate secondary antibodies (Invitrogen). VEGF was detected with 3,3’ diaminobenzidine (DAB) staining. Bone samples were cut at 7-µm sections. Hematoxylin and eosin (H&E) staining, Masson’s trichrome staining, or immunohistochemistry with DAB were performed with standard protocol. Images were collected with Leica DM2500 and QCapture Pro software.
Antibodies and growth factors
As indicated, antibodies included rat monoclonal anti-mouse CD31 (1:50, BD Pharmingen), mouse monoclonal and Cy3-conjugated anti-mouse smooth muscle a-actin SMA (1:200, Sigma), anti-phospho-histone H3 (PH3, Cell Signaling Technology), anti-Flk1 (1:100, Cell Signaling Technology), anti-Flt-1 (1:50, Cell Signaling Technology), anti-mouse F4/80 (1:100, eBioscience), anti-MAC3 (1:100, BD Pharmingen), anti-mouse VEGF (1:50, Neomarkers, Inc). Secondary antibodies included Alexa Fluor® 594-labeled anti-rat or anti-rabbit IgG (1:1000, Jackson Immuno Res) and Alexa Fluor 488-conjugated goat anti-rat IgG (H+L) (Molecular Probes). Growth factors included human recombinant VEGF (VEGF165, R&D Systems), human recombinant FGF-2 (R&D Systems), EGF (Cell Signaling Technology), IGF-1 (Cell Signaling Technology), and IL-3 (R&D Systems).
RT-PCR analysis
Immortalized ECs were generated and cultured as previously described. 22 Total RNA was isolated from tissues or cultured cells using Trizol reagent (Invitrogen) and reverse transcribed into cDNA. Gene expression of the target sequence was analyzed by real-time quantitative (Reverse Transcription- Polymerase Chain Reaction) RT-PCR using the TaqMan or SYBR Master Mix (Roche), analyzed in a StepOnePlus system (Applied Biosystems), and normalized in relation to the expression of 18S ribosomal RNA as an endogenous control. Primers and Taqman probes were designed in Universal Probe Library Assay Design Center from Roche Applied Science. RT-PCR data were analyzed using the ΔΔCt method (threshold values). 26
TaqMan primers:
18S forward primer 5’-AAATCAGTTATGGTTCCTTTGGTC-3’ and reverse primer 5’-GCTCTAGAATTACCACAGTTATCCAA-3’;
VEGF forward primer 5’-GCAGCTTGAGTTAAACGAACG-3’ and reverse primer 5’-GGTTCCCGAAACCCTGAG-3’;
Flk-1 forward primer 5’-CAGTGGTACTGGCAGCTAGAAG-3’ and reverse primer 5’-ACAAGCATACGGGCTTGTTT-3’;
Flt-1 forward primer 5’-GGCCCGGGA TATTTATAAGAAC-3’ and reverse primer 5’-CCATCCATTTTAGGGGAAGTC-3’;
Syber Green primers:
18S forward primer 5’-CGCTTCCTTACCTGGTTGAT-3’ and reverse primer 5’-GAGCGACCAAAGGAACCATA-3’
SDF-1 forward primer 5’-CAGAGCCAACGTCAAGCA-3’ and reverse primer 5’-AGGTACTCTTGGATCCAC-3’;
CXCR-4 forward primer 5’- CCATGGAAC CGATCAGTGTGA-3’ and reverse primer 5’-TTTTCATCCCGGAAGCAGG-3’.
Statistics
Results are expressed as mean value ± SEM. The p-values were calculated using two-tailed unpaired Student’s t-test and considered significant at p ≤ 0.05.
Results
Generation of EC-specific ARNT-deficient adult mice
Recognizing that our ArntloxP/loxP::VeCadherin-Cre+ mice are embryonic lethal (data not shown), 27 we created a mouse genetic system to assess the in vivo requirement of endothelial-ARNT in adult mice by crossing ArntloxP/loxP to inducible VeCadherin-Cre transgenic mice (VeCadherin-CreTM). The efficiency of this Cre mouse was previously reported to result in about 70% deletion in the adult endothelium, whereas there was a low percentage (<0.3%) in BM cells. 21 ArntloxP/loxP::VeCadherin-CreTM+ 8–12-week-old mice were injected with tamoxifen for 10 days and deletion efficiency was assessed by the amplification of a null allele band from genomic tail DNA (ArntΔiEC). 22 This genetic approach inhibits all canonical-HIF transcriptional activity and deletion is limited to the course of tamoxifen injections. At this time, no distinguishable phenotypes under normal physiological conditions were observed in any of the adult animals. All experiments described below were performed 14 days following the start of tamoxifen treatment, thus limiting our in vivo studies to examining the short-term requirements of endothelial-ARNT in adult mice.
Kinetics of cutaneous wound healing are delayed in ArntΔiEC mice
Wound healing is a multifaceted repair mechanism involving coordinated efforts that include inflammatory, epidermal, and vascular cells. We closely evaluated the in vivo consequences to the exclusive loss of endothelial responses to hypoxia by using mice with a stringent postnatal endothelial-specific deletion of ARNT in well-established dermal punch wound-healing assay. We examined the rate of closure on the excisional wounds in three separate experiments and observed a significant decrease during the early time points in the dermal biopsy punches of ArntΔiEC compared to ArntloxP/loxP control mice (Figure 1A). Histological analysis further confirmed visibly larger wounds at day 3 from mutant samples compared to controls. While by day 7 ArntΔiEC wounds were closed with intact epidermis, much granulation tissue remains. In contrast, the epidermis in control mice completely healed with visible hair follicles (Figure 1B). The delayed closure was not due to defects in inflammatory cells since no differences were detected in wounds of ArntΔiEC relative to control mice regarding either the Mac-3+ or F4/80+ myeloid cell numbers (Figures 1C+D and S1A+B). However, while CD31+ staining was observed in day 7 wound sections of control and ArntΔiEC mice, the numbers of mature CD31+/SMA+ vessels were reduced in mutant samples (Figure 1E+F). Furthermore, CD31+/PH3+ cell quantification revealed reduced numbers of proliferating ECs in ArntΔiEC wounds relative to controls (Figures 1G and S1C). While these results confirm reports that HIF is essential in promoting the maintenance, migration, and proliferation of ECs, we hypothesize that reduced infiltration of endothelial progenitor cells as part of the neoangiogenic response may contribute to the delay in wound healing.22,28
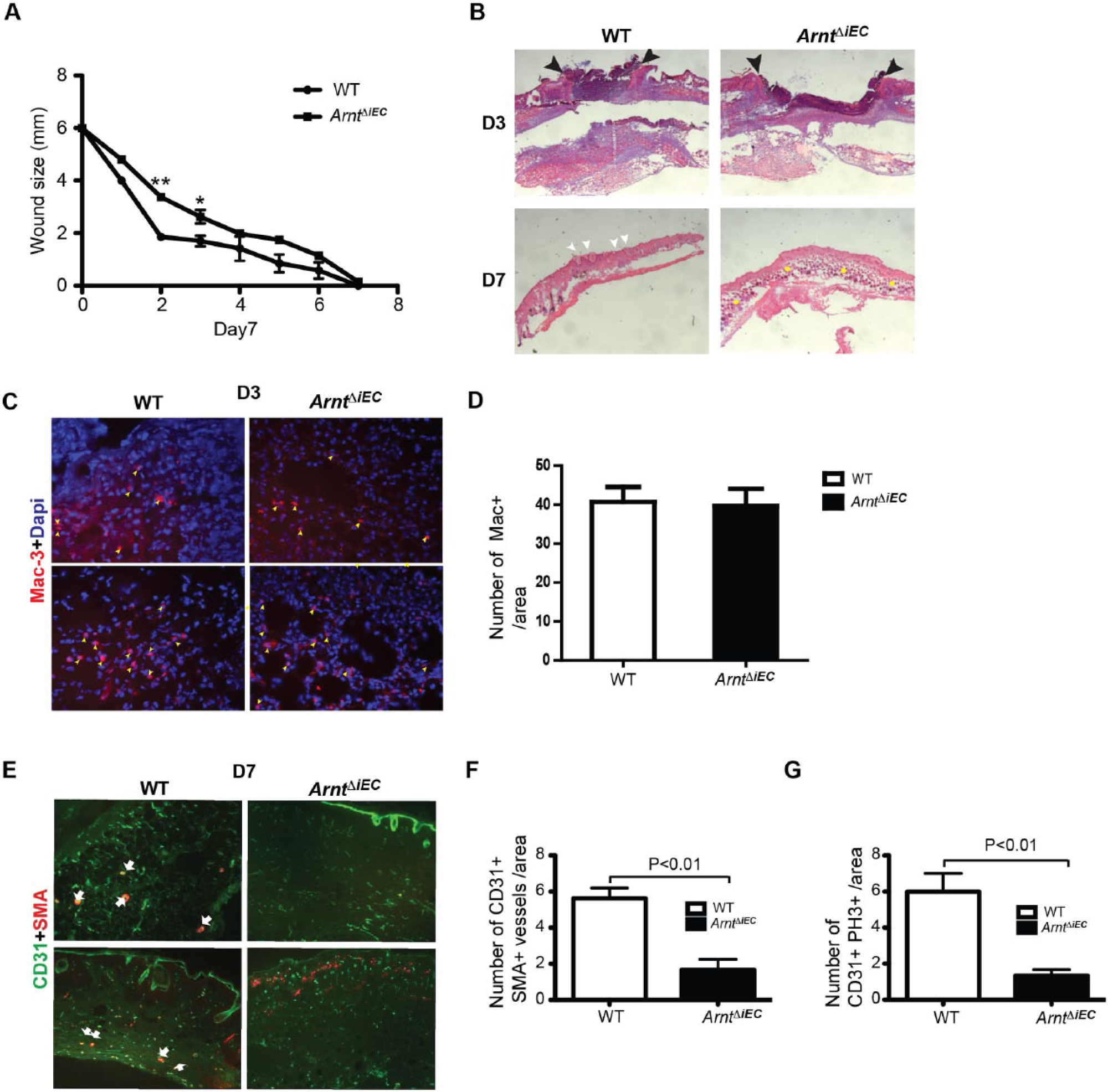
Wound healing is delayed in ArntΔiEC adult mice. Cutaneous wounds (6 mm in diameter) were perforated into the skin of 8-week-old ArntloxP/loxP control and ArntΔiEC mice. (A) The rate of wound closure (n=5) showed a significant delay, especially during the second and third days. Wound widths were plotted against specific time points to determine the rate of their closure. Statistical analysis was performed using the unpaired Student’s t-test, (**p<0.001; *p<0.01). (B) Hematoxylin and eosin (H&E) stained representative sections from days 3 and 7 wounds of control and ArntΔiEC mice. At day 3, mutant wounds were visibly larger (arrowheads) compared to controls. By day 7, wounds of control mice were completely healed and hair follicles regenerated (white arrowheads), but while the ArntΔiEC wounds were closed, they retained much of the granulation tissue (yellow dots). (C) Mac-3 staining of day 3 wound sections indicated that myeloid cell infiltration was not affected by the loss of ARNT in endothelial cells. (D) Quantification of Mac+ cells in day 3 wounds (n=3). (E) Endothelial (CD31, green) and vascular smooth muscle (alpha-smooth muscle actin, red) staining of day 7 wounds showed deficient numbers of mature vessels in ArntΔiEC compared to control mice. Arrows: CD31+/SMA+ vessels. (F) Quantification of CD31+/SMA+ mature vessels in day 7 wounds (n=9). (G) CD31+/PH3+ cell quantification demonstrated reduced numbers of proliferating ECs in ArntΔiEC wounds relative to controls (n=3). (Magnification: panel B, 5×; panel C, 20×; panel E, 10×.)
Matrigel plug in ArntΔiEC mice display reduced migrating cells and fail to form vessel
To further investigate the effects of loss of endothelial-ARNT on endothelial progenitor cell homing and neovascularization in vivo, the mouse Matrigel plug assay was performed.29–31 ArntΔiEC and ArntloxP/loxP control mice were subcutaneously injected with Matrigel containing VEGF and bFGF and recovered after 7 days. First, we confirmed that the plugs resided in a hypoxic microenvironment by examining pimonidazole, an in vivo marker for hypoxia (Figure S2). To visualize vessel integrity, the mice were injected with FITC-dextran prior to euthanasia. Although we observed some CD31+ cell infiltrates in the periphery of the ArntΔiEC Matrigel plugs, the ability to penetrate the core of the implanted plug was significantly reduced (Figure 2A). Quantification of CD31+/dextran+ vessel numbers showed a statistically significant decrease in Matrigel plugs collected from ArntΔiEC mice (five areas per sample, n=3; Figure 2B+C). Considering that Flk-1 is also a critical effector in mobilization of BM-derived endothelial progenitors cells in response to VEGF, Matrigel sections revealed that ArntΔiEC plugs have reduced infiltrating Flk-1+ ECs relative to those within the core of control Matrigel plugs (Figure 2D). The reduced numbers and disturbed integrity of vessels in Matrigel plugs from ArntΔiEC mice further implies that endothelial-ARNT is required for neoangiogenesis and maturation of vessels.
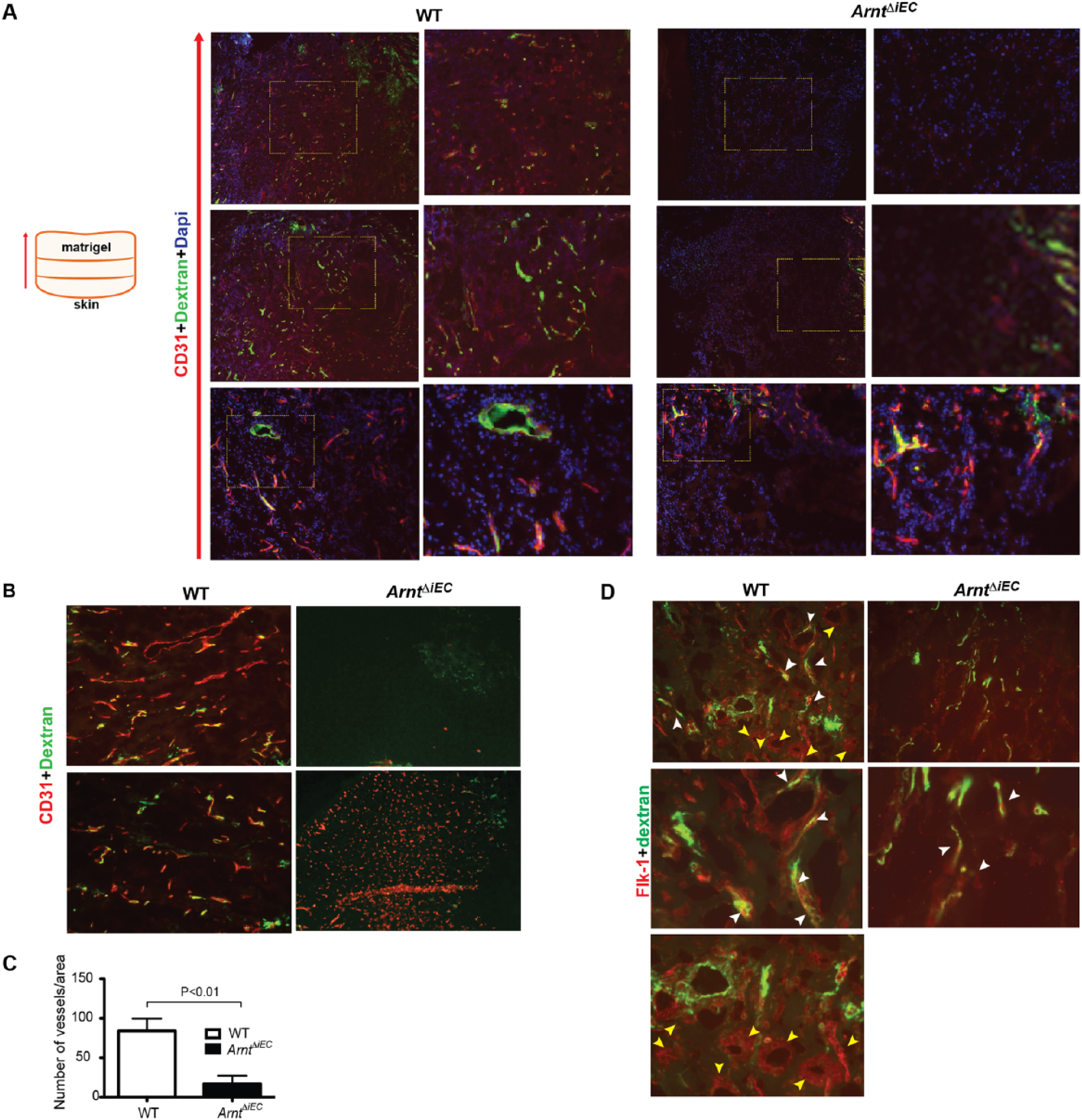
Reduced numbers of infiltrating BMDPPCs and mature vessels in ArntΔiEC Matrigel plugs. (A) 5× and 40× magnification images of CD31 staining (red) and FITC-dextran-infused (green) vessels revealed reduced perfused vessels in ArntΔiEC mice. (B) Note that while some peripheral CD31+ cells were apparent in plugs obtained from mice of either genotype (lower panels), there was a lack of staining in central ArntΔiEC plugs compared to controls (upper panels); 10×. (C) Quantification of intact Dextran+ vessels in Matrigel plugs from control and ArntΔiEC mice (n=5). (D) Overall reduced Flk-1 (red) and Dextran (green) staining in plugs (upper: 20×; lower: 40×). Note the presence of Flk-1+ along with Dextran+ cells (white arrowheads) and the Flk-1+ vascular clusters (yellow arrowheads), potential hemogenic progenitors, exclusive in control samples.
ArntΔiEC mice display decreased BMDPPC mobilization and recruitment in response to exogenous VEGF
The cellular mechanisms resulting in decreased numbers of migrating proangiogenic cells in ArntΔiEC mice may be due to their overall decreased numbers. Since unique markers to these cells have not yet been identified, we considered that our VeCadherin-Cre-inducible system may affect this cell population. We first examined BMDPPC numbers by employing an EPC colony-forming assay (EPC-CFA) methylcellulose method that defines primitive (small, round cell clusters) and definitive (large, spindle-shaped cells) colonies to represent stem/progenitor cells with endothelial properties.25,32 We observed no significant distinction between wild type (WT) and ArntΔiEC samples in numbers of either colony type from Lineage– cells collected from the BM mononuclear cell layer (Figure 3A). When representative colonies are replated onto fibronectin-coated plates, both controls and ArntΔiEC differentiate into ECs defined by their cobblestone appearance and the surface expression of CD31 and Flk-1 (Figure 3B). Thus, it appears that BMDPPCs in this mouse model have no intrinsic differentiation defects.
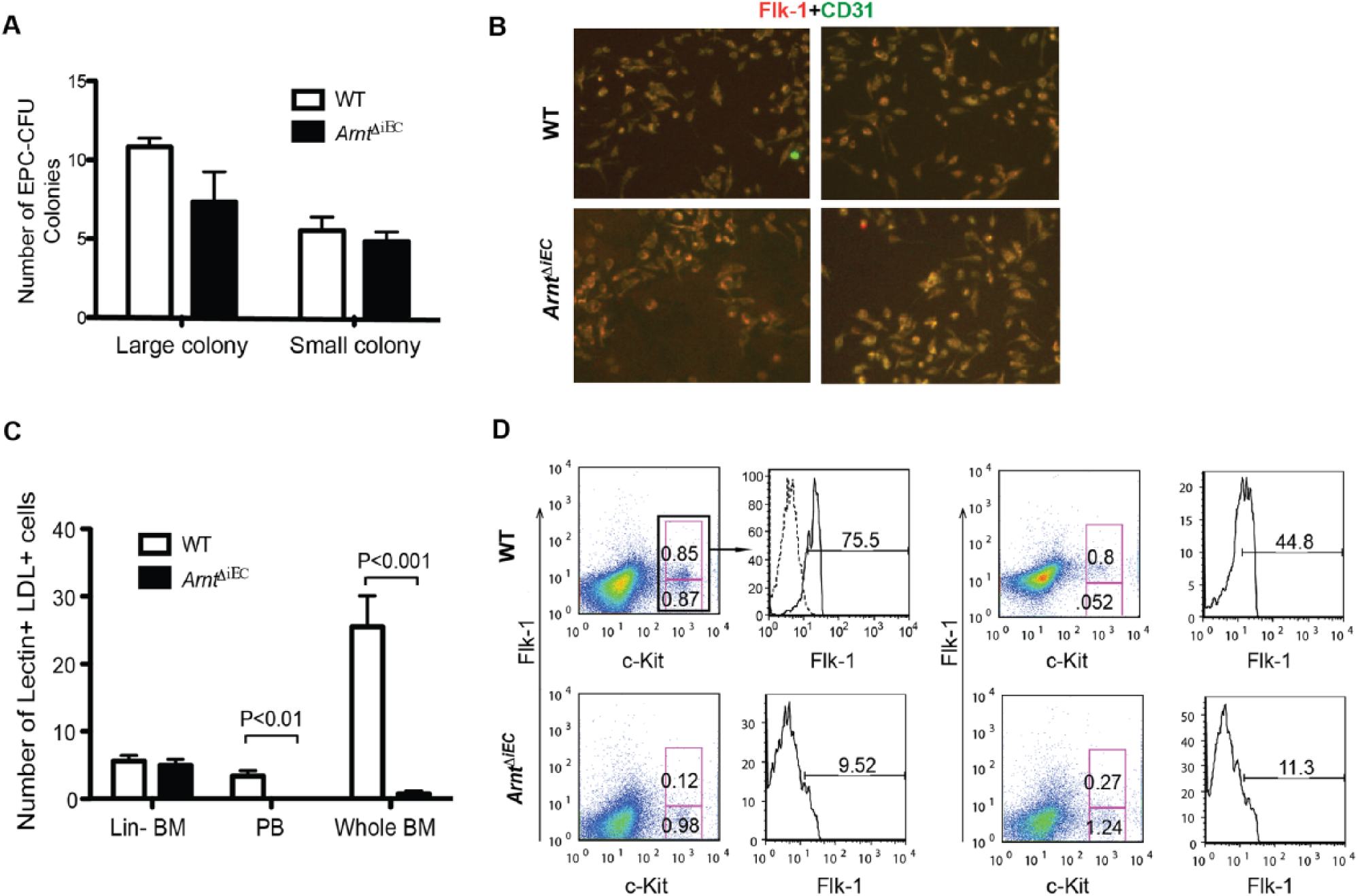
BMDPPCs are reduced in VEGF-treated ArntΔiEC mice. (A) The differentiation profile of EPC-CFU colonies of either ‘definitive’ (large) or ‘primitive’ (small) phenotypes from BM of control and ArntΔiEC mice showed no statistically significant difference. (B) EPC-CFU colonies were replated on fibronectin, allowed to differentiate for 4 days, and stained for Flk-1 and CD31 to corroborate their endothelial progenitor phenotype (20×). (C–D) ArntloxP/loxP and ArntΔiEC mice were treated with rVEGF (100 mg/kg/day) for 4 days and BMDPPCs were analyzed by culturing or flow cytometry. (C) Quantification of DiLDL+ Lectin+ cells indicated overall significantly less numbers of BMDPPCs in either BM or peripheral blood from ArntΔiEC mice. (D) Flow cytometry analysis for the expression of Flk-1 and c-Kit confirmed the decline in numbers of BMDPPCs in BMs of rVEGF-treated ArntΔiEC mice.
Given that VEGF administration stimulates the number of endothelial progenitor cells in the circulation, we hypothesized that BMDPPCs would be reduced in ArntΔiEC mice.29,33 To directly determine if loss of endothelial-ARNT affects BMDPPCs, control and ArntΔiEC mice were subcutaneously injected with rVEGF for 4 days in order to stimulate their mobilization. We first examined the homeostatic state of the BM microenvironment in ArntΔiEC mice following 10 days of our tamoxifen protocol. Histological sections showed no gross BM morphological variances, and immunostaining with CD31 further revealed preservation of the microvasculature in ArntΔiEC and control BM tissues (Figure 4A). In agreement with recent studies demonstrating that VEGF does not affect progenitor cell maintenance within the BM niche, VEGF treatment did not alter the marrow’s gross morphology (Figure 4A).29,34
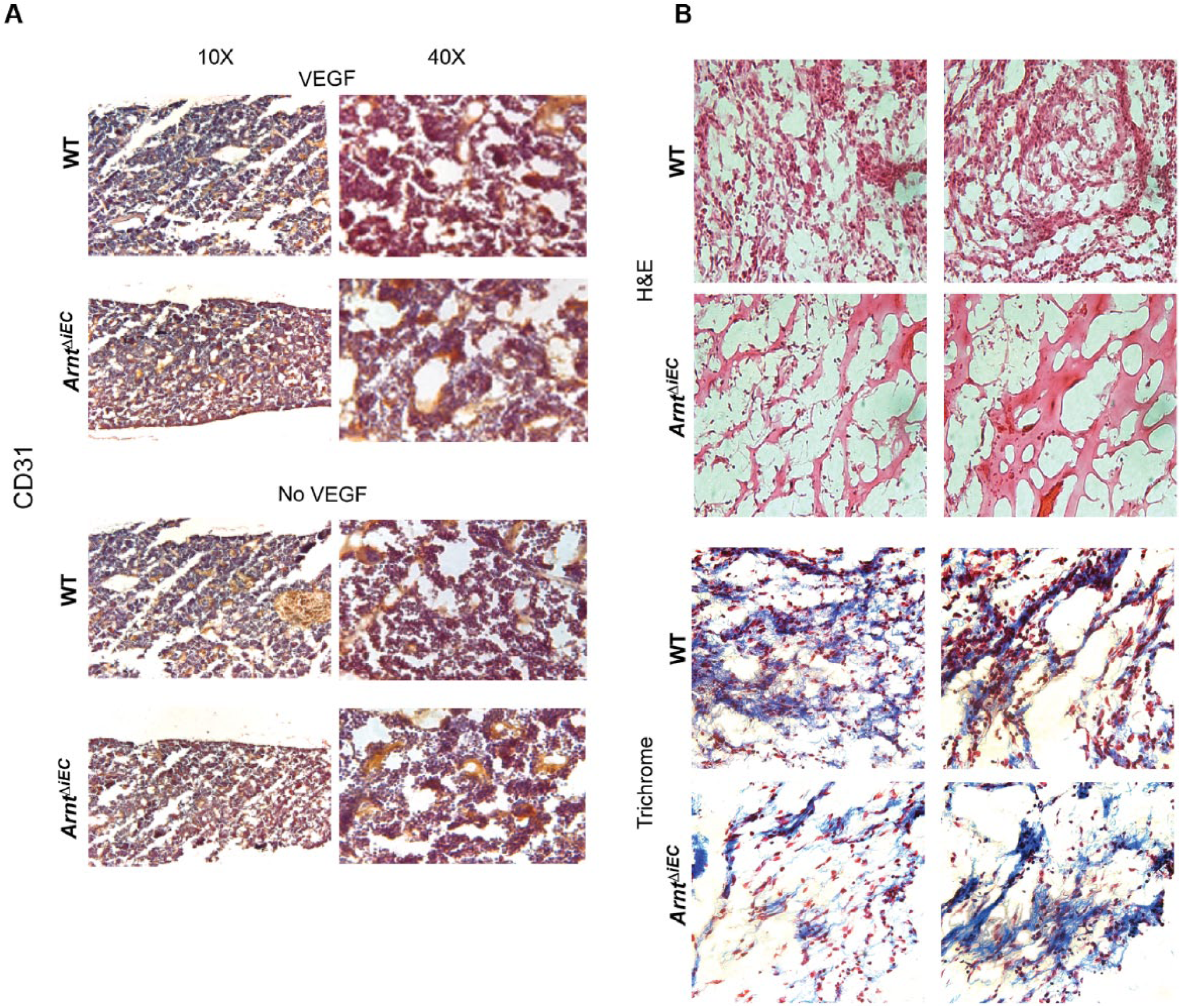
Endothelial-ARNT deletion inhibits the VEGF-induced mobilization of cells into Matrigel plugs. Cohort of control and ArntΔiEC mice were treated with VEGF (100 mg/kg/day) for 4 days and then implanted with Matrigel. (A) No obvious differences were observed in CD31-stained bone marrow sections of non-treated and rVEGF-injected mice. The overall BM cellularity was also not compromised in either control or ArntΔiEC mice. (B) Representative H&E and Masson’s trichrome stained sections of Matrigel plugs showed overall reduced infiltrating cells in rVEGF-treated ArntΔiEC mice compared to controls (20×, n=5).
To examine whether there may be a defect in mobilization of BMDPPC, we took advantage of the EPC culture assay consisting of plating peripheral blood or BM cells directly onto fibronectin-coated plates and enumerating cells uptaking acetylated-LDL and staining with isolectin B4 (Lectin+ LDL+).29,35 By this method, we detected no differences in Lectin+LDL+ cell numbers in Lin– (negative for CD5, CD45R (B220), CD11b, Gr-1 (Ly-6G/C), and Ter-119) BM mononuclear cell (BMMNC) cultures between either cohort of VEGF-treated mice (Figure 3B). On the other hand, plating of unsorted cells resulted in significantly reduced numbers of Lectin+ LDL+ BMDPPC colonies from BM samples collected from ArntΔiEC mice (Figure 3C+D, S3). Noticeably, VEGF treatment resulted in the selective mobilization of BMDPPCs to the peripheral blood of control but not ArntΔiEC mice (Figure 3C+D). This could further explain our in vivo observations that while there were extensive vessels located in Matrigel plugs from rVEGF-treated controls, ArntΔiEC mice had remarkably reduced vascularization in plugs (Figure 4B).
Because there appears to be no intrinsic defects within the selected subpopulation of Lin– BMMNC cells in ArntΔiEC mice as they can effectively generate EPC colonies, we hypothesized that mutant mice have overall reduced proangiogenic progenitor cells. To test this hypothesis, we quantified BMDPPC from BM of rVEGF-treated mice by flow cytometry (Figure 3D). While we did not observe significant overall differences in c-Kit+ cell numbers, there were reduced Flk-1+c-Kithi progenitor cell numbers in BM of ArntΔiEC mice (0.12 and 0.27) compared to controls (values in upper pink gates 0.85 and 0,85 for WT, 0.12 and 0.27 for ArntiΔEC). The percent of Flk-1+ cells from gated c-Kithi were significantly decreased in mutant samples. Collectively, these results argue that the vascular microenvironments in endothelial-HIF-deficient mice inadequately support the maintenance and behavior of BMDPPCs.
Loss of endothelial-ARNT impairs the early presence of migrating CD31+ cells in cutaneous wounds
The compromised closure of cutaneous wounds in ArntΔiEC mice was significantly different during early time points (days 2–3) (Figure 1A). Since after wounding the inflammatory cells (F4/80+ or Mac-3+) were similarly recruited to wounds of WT and ArntiΔiEC mice, we next focused on the early recruitment of neoangiogenic cells. 36
Strikingly, migrating CD31+ ECs were detected in the center of WT wounds by day 1.5, but were absent in the same regions of ArntΔiEC sections (Figure 5A+B). In samples from mice perfused with FITC-dextran, concomitant staining with CD31 were observed in WT vessels in day 3 wounds, but staining was lacking in ArntΔiEC sections, suggesting that the delay in the recruitment of BMDPPCs disrupts the formation of newly remodeled vessels, which further fail to connect to the circulation during this early phase of wound healing (Figure 5C).
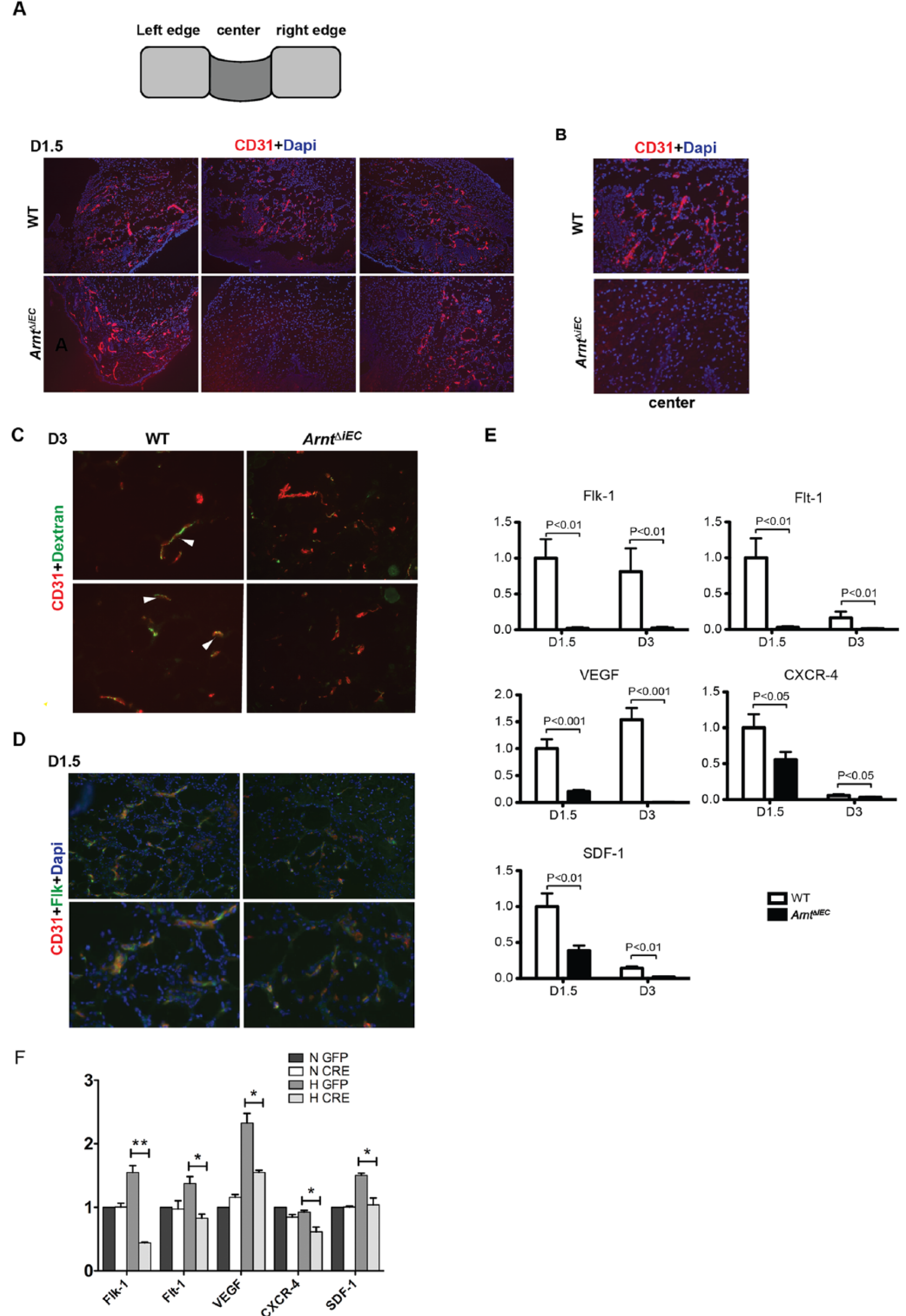
Neovascularization is reduced in ArntΔiEC wound sites. (A, B) Schematic presentation of center and the flanking left and right regions of wound. CD31 staining indicated the lack of infiltrating endothelial cells in the immediate wound area of ArntΔiEC mice at day 1.5 (below, middle panels) at 20× (A) and 40× (B). (C) Representative micrographs of day 3 wound regions from mice-perfused FITC-dextran demonstrated a lack of functional vessels (CD31 (red) / Dextran (green)) in the ArntΔiEC samples. (D) Representative Flk-1 of day 1.5 wounds showed decreased numbers of Flk1+CD31+ECs in ArntΔiEC wounds. (10×). (E) Quantification by real-time PCR revealed a significant decrease of VEGF, Flk-1, Flt-1, SDF-1 and CXCR-4 transcript levels of mRNA isolated from days 1.5 and 3 wound tissues of ArntΔiEC mice relative to controls. (F) Compared to the controls (GFP), transcript levels for Flk-1, Flt-1, VEGF, CXCR-4, and SDF-1 decreased significantly in Arnt-null endothelial cells. (n=3, *p<0.05, **p<0.01.)
It has been postulated that BMDPPCs are involved in the repair mechanisms after endothelial damage and incorporated into sites of neovascularization.6,37 Since clear differences in the migration of proangiogenic CD31+ cells were observed in the early wounds (Figure 5A), we next examined markers of BMDPPC shown to be critical effectors in postnatal neoangiogenesis, including wound-healing processes.25,37,38 Day 1.5 wound sections were stained for Flk-1 (the positive regulator of VEGF signaling), considered an early EPC marker (Figure 5D).34,39,40 Immunochemical analysis of sections from ArntΔiEC wounds show reduced numbers of infiltrating Flk-1+ cells. Mechanistically, these findings are supported by real-time PCR analysis with significantly reduced transcript levels for Flk-1, Flt-1, VEGF, CXCR-4, and, SDF-1 in ArntΔiEC wounds, particularly at day 1.5 (Figure 5E). Real-time analysis using immortalized mouse ECs generated in our laboratory further reveals that under hypoxic conditions, the transcription levels of these tested markers decreased remarkably due to the genetic deletion of ARNT, further suggesting that loss of endothelial-ARNT abrogates neovascularization in dermal wounds by affecting the expression of critical regulators, and thereby recruitment of BMDPPCs (Figure 5F). 22
Loss of endothelial-ARNT impairs the migration of wild-type BM-GFP+ neoangiogenic cells in bone marrow chimeric mice
To further confirm an extrinsic property to this vasculogenic phenotype, blood chimeric ArntΔiEC mice were created by injecting wild-type BM-GFP+ mononuclear cells into sublethally irradiated mice. At day 3 following BM transplantation, Matrigel was again injected into WT and ArntΔiEC chimeric recipient mice, and after 7 days sections were immunostained for GFP and CD31 to assess the recruitment of BM-derived EPC cells to the site of neovascularization. Again, ArntΔiEC Matrigel samples show the overall reduced numbers of CD31+ vessels. While a few, scattered GFP+ cells overlapped with the staining of CD31 vessels throughout the plugs of control mice, most cells appear to stain within vascular lumens, indicating that these were indeed circulating donor cells. In contrast, no GFP+ cells were observed in any sections obtained from ArntΔiEC Matrigel plugs (Figure 6A).
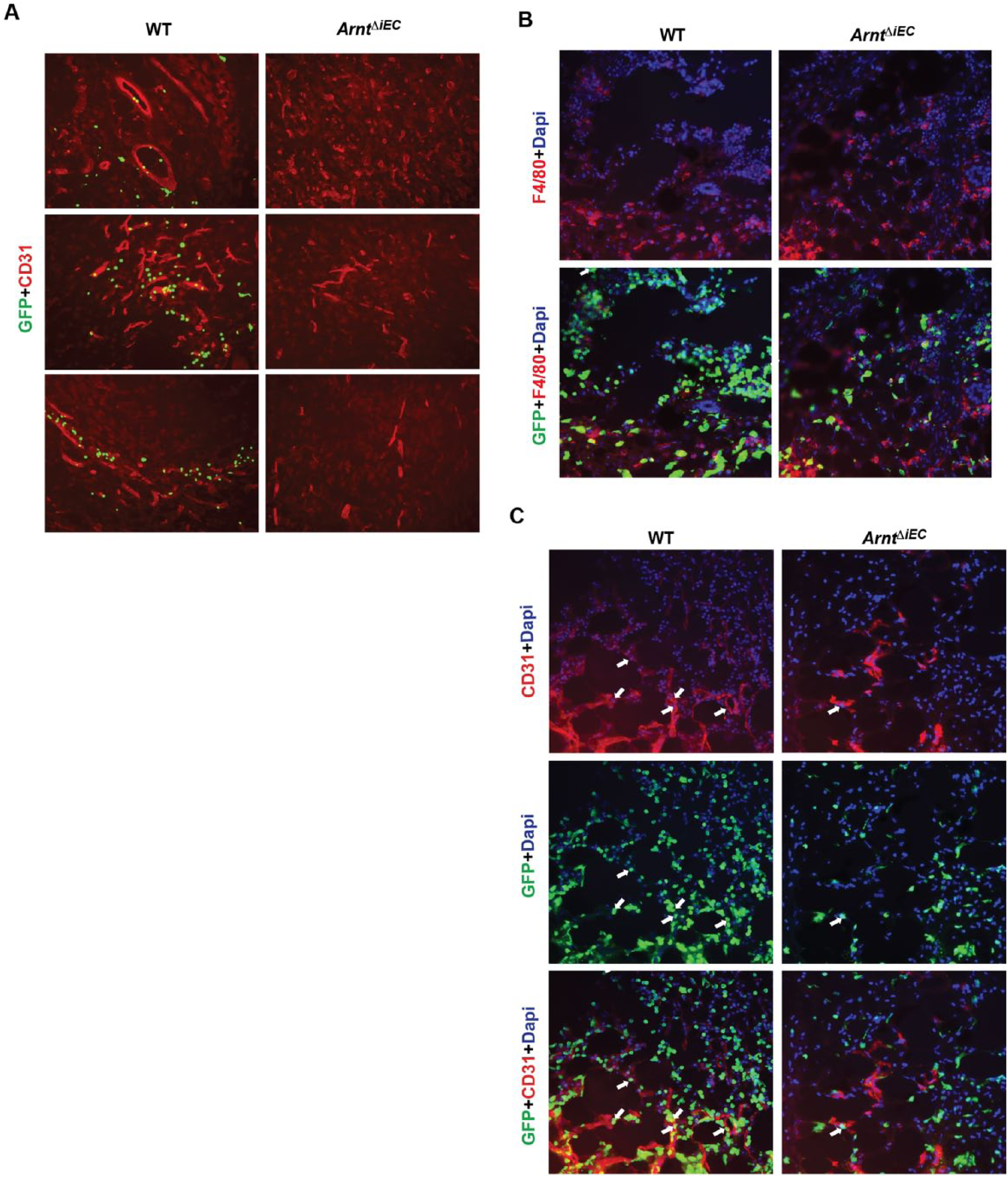
Decreased neovascular contribution of WT GFP+ cells in ArntΔiEC bone marrow chimeric mice. (A) Three days following transplantation of WT GFP+ BM cells (1×106) into sublethally irradiated mice, Matrigel gel was injected and then harvested at day 7. Donor bone marrow cells failed to be incorporated into ArntΔiEC Matrigel plugs. (B, C) Representative micrographs of day 3 wounds indicated a reduction of GFP+ infiltrating cells in ArntΔiEC wound sites. (B) F4/80 (red) costaining showed a similar contribution to wounds in either WT or ArntΔiEC chimeric mice. (C) Compared to controls, the numbers of BM-derived WT GFP+/CD31+ cells (arrows) decreased significantly in ArntΔiEC wounds.
A distinct set of mice with warranted engraftment of GFP+ cells was then generated to create highly chimeric BMs. Recipient mice were sustained for 4 weeks following 7 Gy and two injections of 5 million BMMNCs before performing wound-healing experiments. Immunostaining of day 3 wounds demonstrated significantly less GFP+ cells in ArntΔiEC samples (Figure 6B+C). Moreover, although there was no difference between control and ArntΔiEC regarding the infiltration of F4/80+ myeloid cells (Figure 6B), consistent with our initial observations, overall CD31+ as well as GFP+/CD31+ cell numbers were less in ArntΔiEC wounds (Figure 6C). Collectively, our results suggest that endothelial-HIF is involved in the recruitment and/or migration of BMDPPCs.
Discussion
We show here that ArntΔiEC mice have impaired vasculogenic responses. Specifically, we observed decreased recruitment of BMDPPCs in ArntΔiEC mice utilizing in vivo neoangiogenic models of wound healing and chemokine-induced (soluble or in Matrigel) cell migration. ArntΔiEC mice had reduced numbers of BMDPPCs in the BM and peripheral blood following rVEGF injections. Also, histological analysis showing a lack of WT GFP+CD31+ cells in early wounds of the chimeric ArntΔiEC mice argues that the neoangiogenic phenotype is not intrinsic to BMDPPCs but to the signals provided by the endothelium. Taken together, these experiments strongly argue that HIF transcriptional activity in ECs affects the recruitment of the BMDPPCs required for neoangiogenesis, in part by modulating the expression of chemokines that include stromal cell-derived factor (SDF) 1α and VEGF.
The restricted inactivation of endothelial-ARNT in adult mice significantly dampened endothelial-controlled vascular responses consistent with previous reports showing requirement for proper responses to hypoxia. 41 Importantly, this 2-week in vivo temporal loss of endothelial-ARNT does not appear to affect vessel homeostasis, but a significant scarcity of ECs in ring explants, Matrigel plugs, or dermal wounds in experimental tissues present additional evidence that HIF-transcriptional activity in ECs plays a direct role during adult vascular responses. 22 We and others have shown that HIF-deficient ECs display adverse defects in migration, proliferation, and survival that may also contribute to the observed postponement of wound healing in ArntΔiEC mice.13,22,42 These experiments also strongly support that HIF transcriptional activity in vascular ECs is critical for multiple cellular functions necessary for the regulation of the vascular endothelium. Overall, impaired HIF-signaling exclusively in the endothelium of adult mice affects the recruitment of BMDPPCs in addition to the remodeling of vessels.
HIF is considered to play a critical role in various aspects of wound healing. 43 Specifically, BM-derived angiogenic cells (BMDACs) play an important role in neovascularization, and an overall compromised healing response in burned wounding of heterozygous Hif1-α mice is partially due to their decreased mobilization.31,44 While our key observation that the ArntΔiEC mice wound phenotype results in an early delay and reduced numbers of migrating BMDPPCs is consistent with a recent report of a mouse model with loss of HIF-1α or ARNT activity in Tie2+ cells, the model has distinct features. 45 First, punch and burn wounds produce different healing responses whereby thermal wounds create overall greater necrosis and destruction of vessels. Second, the present analyses explored the consequences resulting from the temporal and restricted deletion of ARNT using inducible VecadCreERT2 mice excluding potential secondary developmental or sustained angiogenic defects. In contrast, disruption of Arnt utilizing Tie2-Cre recombination results in 90% embryonic lethality. 27 Third, recombination withTie2-Cre is also highly active in BM hematopoietic precursor cells and affecting myeloid and lymphoid cells.46,47 Indeed, this Tie2-Cre genetic mouse model generated intrinsic defect BMDACs contributing to the reported reduced numbers of circulating CXCR-4+/Tie2+ cells. 44 Our present study indicates that there appears to be no intrinsic defects to BMDPPCs, but rather that the endothelial inadequacy of HIF is responsible for the aberrant numbers of BMDPPCs that result in neoangiogenic deficiencies. Our data strongly suggest that EC-HIF extrinsic global effects are critical to vascular biology.
The ineffective vessel dynamics in ArntΔiEC mice suggests an important mechanism by which HIF-deficient vessels are unable to promote neoangiogenesis in response to an ischemic microenvironment. SDF-1 has a significant effect on the EPC recruitment as a consequence of ischemia. 18 The recruitment of EPCs to ischemic tissue has been shown to be proportional to the expression of SDF-1, primarily produced by ECs, and as a consequence of HIF-1 expression. 17 Our molecular data shows that Arnt-null ECs fail to induce the expression of Vegf and Sdf-1 in response to hypoxia, while the specific inactivation of canonical HIF in the endothelium of 8-week-old ArntΔiEC mice also results in reduced SDF-1 expression. Their inability to respond to the physiological hypoxic trigger generated by injured tissues led to the inefficient participation of EPCs, by affecting the important regional expression of SDF-1 required for their proper recruitment. The inefficiency of triggering this angiocrine pathway is consistent with Ceradini et al., demonstrating that the specific blockade of SDF-1 or CXCR-4 reduces the engraftment of EPCs into ischemic tissues. 17
In ArntΔiEC wounds, we observed decreased expression of genes involved in two pathways. One disrupted pathway is VEGF and its receptors Flk-1 and Flt-1, while the second is SDF-1 and its receptor CXCR-4, transcripts previously shown to be induced by hypoxia. These chemokine pathways have been shown to be involved in the mobilization of EPCs.34,36,48–51 In addition, EPCs are susceptible to hypoxic gradients. 52 The altered biochemical environment resulting from loss of HIF transcriptional activity in the endothelium of wounds could contribute to the inadequate migration of BMDPPCs during the early phases of wound healing, as has been indicated in aging patients and mice.44,53,54 Expression of SDF-1 and HIF-1 in response to burn wounding has been shown to be impaired in aging mice. 53
A growing number of reports demonstrate EPCs’ importance during endothelial repair and vascular homeostasis including myocardial ischemia, retinopathy, and wound healing.55–58 EPC number and function are associated with the severity of peripheral artery disease (PAD) and their numbers can predict cardiovascular outcomes in PAD patients. 59 Mobilization and homing of these cells is attenuated in older patients or those suffering from various diseases including perivascular pathologies.60–65 Specifically, failure of aging tissues to activate the HIF may be associated with the dampened ability for EPCs to circulate.44,54 The present observations have important relevance to these clinical settings as we speculate that the ineffective responses by HIF-deficient endothelium may lead to accelerated vascular aging thereby impacting BMDPPCs, neovascularization, and peripheral vascular diseases. These investigations demonstrating that loss of endothelial-HIF results in reduced BMDPPCs reflect results obtained from clinical studies and lay a foundation for generating novel therapies that promote targeting the mobilization of BMDPPCs in order to promote vessel growth in certain human pathologies.
Footnotes
Declaration of conflicting interest
The authors declare that there is no conflict of interest.
Funding
Technical support was provided by NIH-supported core facilities of the Case Comprehensive Cancer Center (CA43703). This work was supported by the National Heart, Lung, and Blood Institute R01-HL096597 (DRB).