
Abstract
The incidence of metabolic syndrome is rapidly increasing in the United States and worldwide. The metabolic syndrome is a complex metabolic and vascular disorder that is associated with inappropriate activation of the renin–angiotensin–aldosterone system (RAAS) in the cardiovascular (CV) system and increased CV morbidity and mortality. Insulin activation of the phosphatidylinositol-3-kinase (PI3K) pathway promotes nitric oxide (NO) production in the endothelium and glucose uptake in insulin-sensitive tissues. Angiotensin (Ang) II inhibits insulin-mediated PI3K pathway activation, thereby impairing endothelial NO production and Glut-4 translocation in insulin-sensitive tissues, which results in vascular and systemic insulin resistance, respectively. On the other hand, Ang II enhances insulin-mediated activation of the mitogen-activated protein kinase (MAPK) pathway, which leads to vasoconstriction and pathologic vascular cellular growth. Therefore, the interaction of Ang II with insulin signaling is fully operative not only in insulin-sensitive tissues but also in CV tissues, thereby linking insulin resistance and CV disease. This notion is further supported by an increasing number of experimental and clinical studies indicating that pharmacological blockade of RAAS improves insulin sensitivity and endothelial function, as well as reduces the incidence of new-onset diabetes in high-risk patients with CV disease. This article reviews experimental and clinical data elucidating the physiological and pathophysiological role of the interaction between insulin and RAAS in the development of insulin resistance as well as CV disease.
Introduction
Diminished tissue sensitivity to insulin is a characteristic feature of various pathological conditions termed the metabolic syndrome, also known as the cardiometabolic syndrome. Factors that contribute to the complex interaction between genetic and environmental factors underlying impaired insulin signaling include obesity, physical inactivity, and aging. 1 Clinically, the cardiometabolic syndrome is characterized by central obesity, hypertension, hyperlipidemia, hyperinsulinemia, microalbuminuria, and endothelial dysfunction, 2 all of which are risk factors for cardiovascular (CV) and chronic kidney diseases.2,3 Insulin resistance is central to the pathophysiology of the cardiometabolic syndrome. Increasing evidence demonstrates that insulin resistance can develop not only in tissues classically responsive to insulin, but also in CV tissues, where it participates in the development of CV disease.4,5
The cardiometabolic syndrome is a complex metabolic and vascular syndrome that is associated with CV activation of the renin–angiotensin–aldosterone system (RAAS) and oxidative stress as well as a substantial risk for CV and chronic kidney diseases. 5 RAAS is an endocrine system predominantly responsible for the regulation of systolic blood pressure as well as salt and water homeostasis. Angiotensin (Ang) II, however, has many actions beyond its effects on blood pressure and has been shown to inhibit insulin signaling.6,7 Indeed, insulin and Ang II are two important hormones in the control of metabolic and hemodynamic homeostasis, respectively. Functional interaction between Ang II and insulin is operative in insulin-sensitive tissues as well as the CV system, and participates in the regulation of metabolism and CV function.6,8,9 The evidence of a link between insulin resistance and an inappropriately overactive renin–angiotensin system (RAS)has been implicated in the pathogenesis of hypertension, obesity, and type II diabetes mellitus (T2DM).10,11 A growing number of experimental and clinical studies12,13 have provided evidence indicating that pharmacological blockade of the RAAS by either angiotensin-converting enzyme (ACE) inhibitors or angiotensin II type 1 receptor blockers (ARBs) not only reduces CV injury but also improves insulin sensitivity and reduces the incidence of new onset T2DM in subjects with hypertension and/or CV disease, independently of blood pressure-reducing effects.
This review focuses on: (1) the CV actions of insulin; (2) the association between inappropriate activation of RAAS and insulin resistance in the development of CV diseases; and (3) experimental and clinical evidence supporting the role of RAAS blockade in the prevention and treatment of metabolic and CV diseases.
Cardiovascular actions of insulin
Insulin has important vascular effects that contribute to either vascular protection or injury (Figure 1). 14 In the vascular endothelium, insulin stimulates two major signaling transduction cascades: the phosphatidylinositol-3-kinase (PI3K) and the mitogen-activated protein kinase (MAPK) pathways. 15 Among the most important CV actions and vasoprotective effects of insulin is the stimulation of nitric oxide (NO) production in the endothelium. 16 It is clear that insulin stimulates endothelial NO production via activation of the PI3K pathway, which phosphorylates and activates Akt, which in turn phosphorylates and activates endothelial nitric oxide synthase (eNOS), resulting in increased NO production. 16 On the other hand, insulin also promotes a host of deleterious vascular effects by stimulating the actions of various growth factors acting through stimulation of the MAPK pathway, leading to vasoconstriction and proatherogenic effects. 4
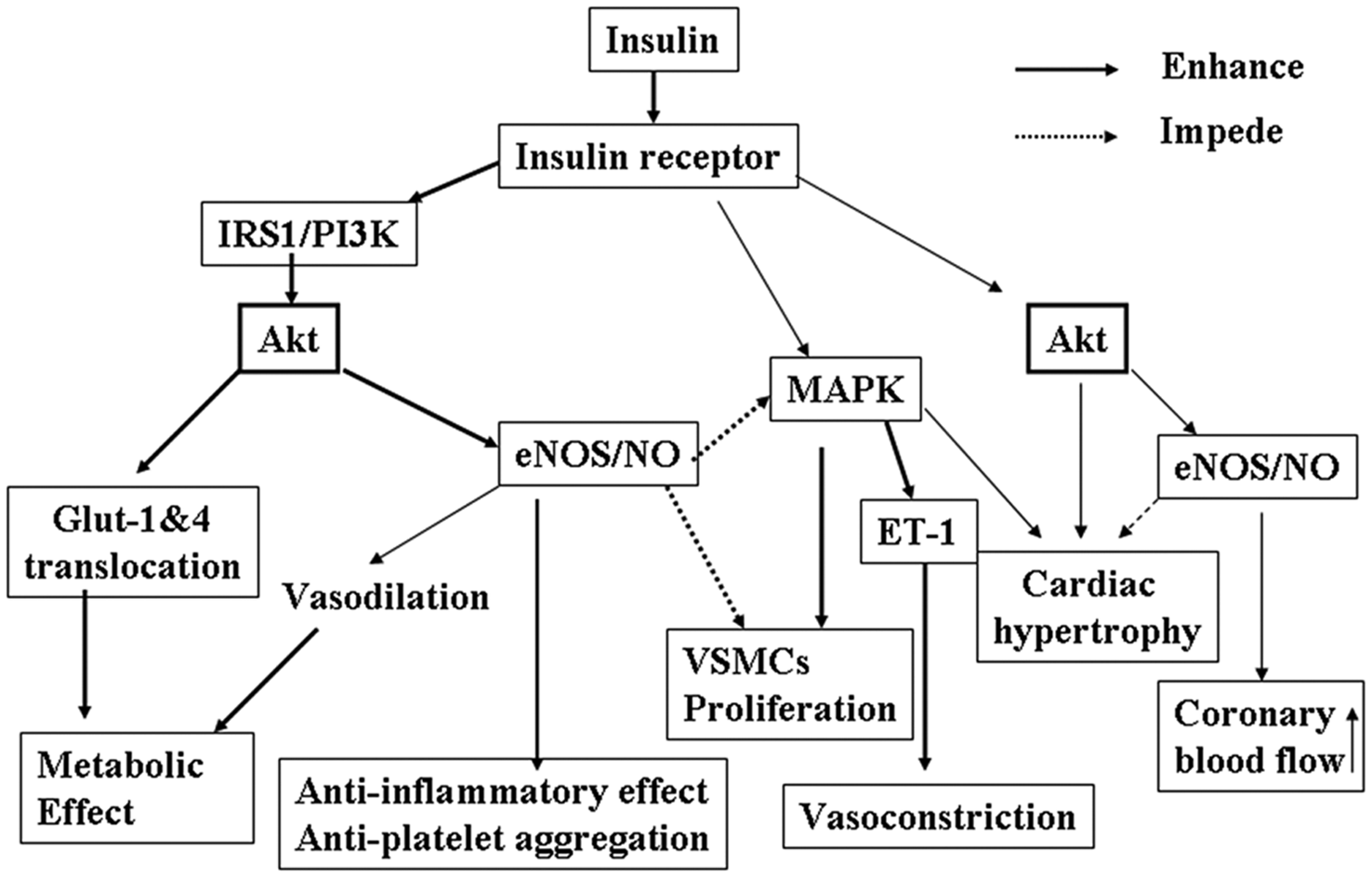
Insulin actions and insulin signaling pathways in the cardiovascular system. Insulin, after binding its receptor, stimulates two signaling cascades: insulin receptor substrate-1 (IRS-1)/phosphatidylinositol 3-kinase (PI3K)/Akt and mitogen-activated protein kinase (MAPK). Downstream events of phospho-Akt include: (1) stimulation of Glut-1 (vascular cells) or Glut-4 (skeletal muscle, liver and fat tissues) translocation into membrane, which mediates insulin’s metabolic effects; and (2) phosphorylation of endothelial nitric oxide synthase (eNOS) and production of nitric oxide (NO), which induces vasorelaxation and anti-inflammatory and antiatherogenic effects. Insulin activation of MAPK promotes vascular smooth muscle cell (VSMC) proliferation/migration and release of endothelin-1 (ET-1), which induces pro-atherogenic and vasoconstriction effects. In the heart, insulin increases coronary blood flow through stimulation of eNOS/NO production and promotes cardiomyocyte hypertrophy through phosphorylation of Akt and activation of MAPK.
Hemodynamic actions of insulin
In the CV system, insulin contributes to hemodynamic regulation by multiple mechanisms. 14 Vasodilation is one of the most important vascular actions of insulin. It has been shown that insulin induces vasodilation in arteries and veins, leading to increased microcirculatory flow via increased capillary recruitment, through stimulation of endothelial NO production.12,16 For instance, insulin infusion at higher physiological concentrations under euglycemic glucose clamp conditions causes a dose-dependent increase in blood flow to skeletal muscle. 17
Vasodilator responses to insulin-stimulated NO production comprise two distinct stages. First, insulin dilates the terminal arterioles to increase capillary recruitment without concomitant changes in total limb blood flow. This is subsequently followed by relaxation of larger resistance arteries and leads to increased limb blood flow. 18 Animal and human studies have shown that vasodilation leading to increased blood flow in the skeletal muscle is a major physiological consequence of the insulin-stimulated PI3K-eNOS pathway, leading to increased NO production in the endothelium, which may contribute to increased glucose disposal. 16 The NO-dependent vasodilator effects of insulin are antagonized by the vasoconstrictor effects of insulin mediated by activation of the sympathetic nervous system and stimulation of the secretion of the vasoconstrictor endothelin-1 (ET-1) in the vascular endothelium.19,20 In healthy humans, the acute physiological and pharmacological concentration of insulin increases venous plasma catecholamine concentrations and sympathetic outflow in the skeletal muscle.12,19 Another biological action of insulin that may impact hemodynamic homeostasis is stimulation of ET-1 release from the endothelium. 20 ET-1 is a potent vasoconstrictor. Insulin induces ET-1 release via stimulation of the MAPK pathway, as evidenced by the finding that MAPK inhibition blocks the vasoconstrictor effects of insulin. 20 Thus, insulin exerts opposing vasodilator and vasoconstrictor actions with negligible net effect on blood pressure in normal individuals. However, in states of insulin resistance, such as obesity and T2DM, there is impairment in insulin-stimulated NO production and activation of the sympathetic system due to chronic hyperinsulinemia, which shifts the balance in favor of vasoconstriction, thereby promoting hypertension and CV disease. 12
Anti-inflammatory and atheroprotective effects of insulin
It has been shown that insulin at physiological concentrations has anti-inflammatory and anti-atherogenic effects via stimulation of NO production in the endothelium. 21 NO is an important vasoprotective molecule and the endothelium is the first organ that insulin encounters after it is secreted into the circulation. It has been shown that NO induces vasodilation and inhibits vascular smooth muscle cell migration and proliferation, inflammatory cell infiltration into the vascular wall, and platelet aggregation, all of which contribute to the atheroprotective effect of insulin. 22 Therefore, constitutive stimulation of NO production by insulin may play a crucial role for maintenance of vascular health under physiological conditions. Indeed, recent evidence shows that insulin exerts vasodilatory, anti-platelet and anti-inflammatory effects at the cellular level in vitro and in vivo.23–25 In Apo E-deficient mice, insulin administration reduced the number and the size of atherosclerotic lesions. 26 Infusions of low doses of insulin to obese subjects significantly reduced reactive oxygen species (ROS) and plasma levels of inflammatory cytokine markers, including intracellular adhesion molecule-1 (ICAM-1), monocyte chemoattractant protein-1 (MCP-1), and plasminogen activator inhibitor-1 (PAI-1).21,27 Furthermore, a low dose of insulin infusion that did not lower plasma glucose concentration markedly improved clinical outcomes in patients with acute myocardial infarction, accompanied by a significant reduction in plasma C-reactive protein, an index of systemic inflammation. 28
Heme oxygenase (HO)1 is an important antioxidant molecule that has been shown to protect against oxidative injury in atherogenesis.29,30 Insulin increases HO1 expression in vitro and in vivo through activation of the PI3K/Akt pathway.31,32 Moreover, insulin upregulation of HO1 prevented H2O2-induced nuclear factor-κB (NFκB) and caspase-8 activation and apoptosis in pericytes. 31 Therefore, upregulation of HO1 may be another mechanism underlying the anti-inflammatory and atheroprotective effects of insulin.
Proinflammatory and proatherogenic effects of insulin
Insulin is a growth factor that regulates several proto-oncogene transcriptional factors, including c-fos and MAPK. 33 The growth-stimulating effects of insulin may importantly contribute to increased CV risk. Obese individuals and those with T2DM are at a greater risk of developing atherosclerosis than the rest of the population. 34 The underlying mechanisms may involve compensatory hyperinsulinemia in these subjects. 35 Evidence from human and animal studies have shown that compensatory hyperinsulinemia resulting from insulin resistance stimulates expression of PAI-1, vascular cell adhesion molecule-1 (VCAM-1), and E-selectin via a MAPK-dependent mechanism, which may contribute to accelerated atherosclerosis. 36 In addition, recent studies have shown that insulin increases expression of arterial angiotensinogen and angiotensin type 1 (AT1) receptor in cultured vascular smooth muscle cells.37,38 Increased AT1 receptor by insulin may be another mechanism by which hyperinsulinemia promotes the development of CV diseases in obese and type II diabetic individuals with insulin resistance.
Regulation of cardiac function by insulin
Insulin participates in the regulation of cardiac metabolism, growth, contractility, and coronary blood flow. Insulin regulates metabolism in the heart by modulating glucose transport, glycolysis, glycogen synthesis and lipid metabolism. 14 A normal heart derives approximately 70% of its energy from fatty acids, whereas the failing heart shifts its metabolic profile to glucose, instead of fatty acids, as the primary energy substrate. 39 The metabolic actions of insulin are mediated through a PI3K-dependent pathway, which stimulates Akt phosphorylation and Glut-4 translocation to the membrane, thereby facilitating glucose uptake. 40 Myocardial insulin resistance involving the metabolic pathway results in a number of changes in the signaling cascade that lead to inhibition of insulin-mediated phosphorylation of insulin receptor substrate-1 (IRS-1) tyrosine residues and a specific impairment of the PI3K pathway. 40 Signaling through the IRS-1/PI3K/Akt pathway is also essential for developmental and physiological growth of the heart. 14 Constitutive overexpression of Akt leads to cardiac hypertrophy and dysfunction. Pathological cardiomyocyte hypertrophy is regulated by a subset of signaling pathways including insulin activation of PI3K/Akt and MAPK pathways. 41 Insulin increases cardiac contractility through activation of L-type Ca2+ channels, leading to increased Ca2+ influx into cardiac myocytes. 42 Insulin enhancement of cardiac contractility increases myocardial work and oxygen consumption, resulting in a compensatory increase in coronary blood flow. In healthy subjects, insulin enhances myocardial blood flow and decreases coronary vascular resistance. The underlying mechanisms involve stimulation of NO production in the endothelium. 14
Activation of RAAS
RAAS plays an important role in the regulation of physiological processes of the CV system. It is an enzymatic cascade that starts with the cleavage of angiotensinogen by renin to form the inactive decapeptide Ang I. Thereafter, Ang I is converted by ACE to form Ang II. Alternatively, a recently identified carboxypeptidase, ACE2, cleaves one amino acid from either Ang I or Ang II, decreasing Ang II levels and increasing Ang 1–7, 43 which has vasodilator properties. Thus, the balance between ACE and ACE2 is an important factor regulating Ang II levels. The major biologically active products of the RAAS are Ang II and aldosterone. Ang II acts on both AT1 and AT2 receptors. The AT1 receptor is ubiquitously and abundantly distributed in adult tissues, including blood vessels, heart, kidney, adrenal gland, liver, brain, and lung. The AT1 receptor mediates all the classic and well-known effects of Ang II, such as elevation of blood pressure, vasoconstriction, increase in cardiac contractility, aldosterone release from nerve endings, an increase in renal sodium and water reabsorption, and Ang II-induced growth in CV and renal tissues. 22 Aldosterone is a steroid hormone with mineralocorticoid activity that is produced mainly by the adrenal glomerulosa in response to Ang II. The best known physiological role of aldosterone is to increase sodium reabsorption in the distal nephron to maintain sodium balance via activation of the apical epithelial sodium channel and the basolateral Na+, K+-ATPase. Beyond its effects on sodium reabsorption, aldosterone exerts effects on the kidney, blood vessels and the heart, which can have pathophysiological consequences. 44 The circulating RAAS participates in the short-term regulation of the CV system. It becomes activated in acute conditions, including hypotension, hypovolemia, and hemorrhage. In chronic conditions, including hypertension and heart failure, activation of the RAAS participates in the long-term regulation of CV homeostasis via sustained activation of local angiotensin and degradation of bradykinin, resulting in permanent CV structural changes.22,45
Upregulation of RAAS has been found in the CV system in T2DM individuals, which may contribute to the development of many diabetic CV complications, including microvascular and macrovascular diseases. 4 Hyperglycemia increases transcription of angiotensinogen and Ang II production from the local ACE. Obesity, a common cause of insulin resistance, is associated with upregulation of RAAS in adipose tissue, which may trigger chronic inflammatory processes, resulting in insulin resistance.46,47
The interaction of RAAS and insulin signaling in the CV system
Insulin and Ang II are two important hormones in the control of metabolic and hemodynamic homeostasis, respectively.7,8 Recent studies have suggested that the signal transduction pathways of insulin and Ang II share a number of downstream effectors and cross-talk at multiple levels 6 (Figure 2). Insulin signaling is initiated by circulating insulin binding to its receptor. The insulin receptor is a heterotetrameric tyrosine kinase that after binding insulin undergoes a rapid tyrosine autophosphorylation that activates the receptor kinase and allows transient interaction with IRS-1. Interaction of tyrosine phosphorylated IRS-1 with PI3K results in PI3K activation and Akt phosphorylation, which stimulates translocation of Glut-4 to the sarcolemma to facilitate glucose uptake and NO production in the endothelium to induce vasorelaxation. 17
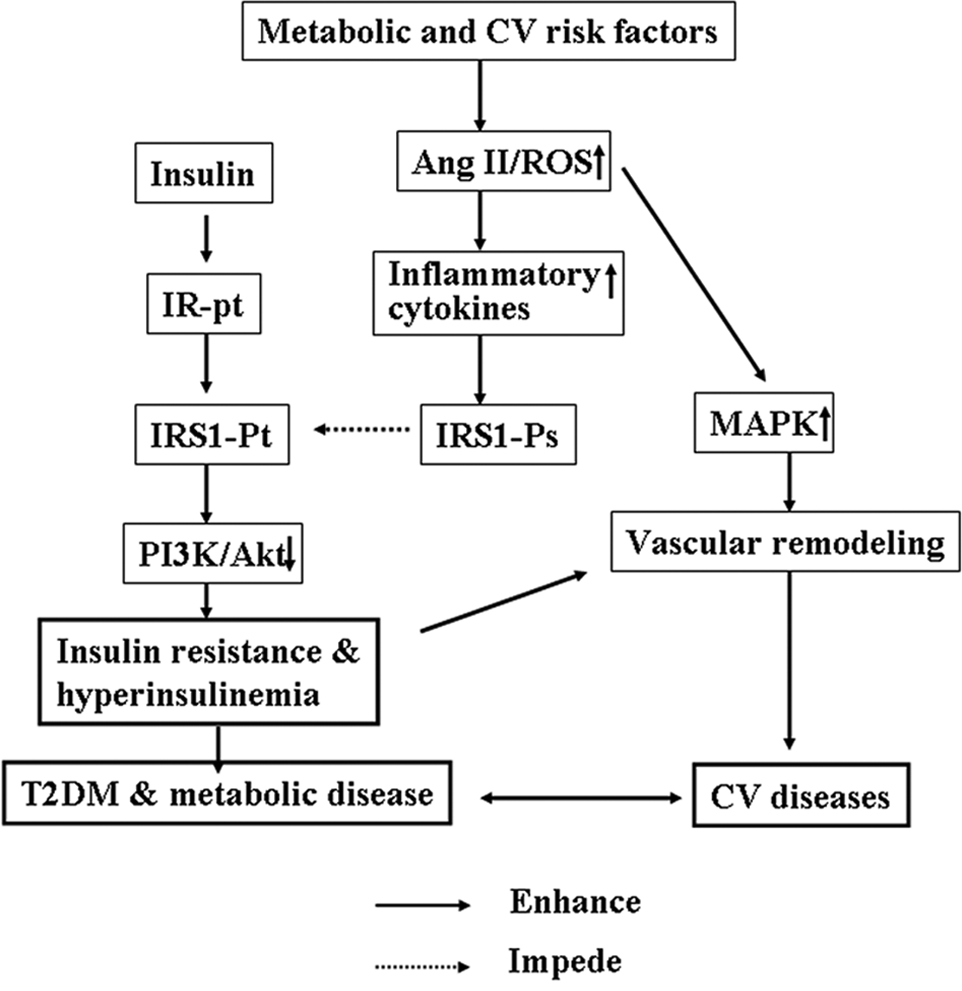
Interaction of insulin and angiotensin (Ang) II in cardiovascular (CV) and metabolic diseases. Metabolic or CV risk factors stimulate the renin–Ang system to increase production of reactive oxygen species (ROS). ROS activate redox-sensitive inflammatory signaling pathways such as nuclear factor κB to increase production of proinflammatory cytokines, which induce phosphorylation of insulin receptor substrate-1 (IRS-1) at serine residues (Ps), then inhibit insulinstimulated phosphorylation of IRS-1 at tyrosine residues (Pt), and consequently inhibit insulin signaling through the phosphatidylinositol 3-kinase (PI3K) pathway, resulting in insulin resistance, hyperinsulinemia, type II diabetes mellitus (T2DM) and metabolic diseases. On the other hand, Ang II/ROS activates the mitogen-activated protein kinase (MAPK) pathway to induce vascular remodeling, which promotes CV disease.
Ang II has been shown to inhibit the insulin-PI3K signaling pathway in both vascular and skeletal muscle cells.6,48 In cultured L6 myocytes, Ang II stimulates serine-phosphorylation of IRS-1. 49 IRS-1 is one of the major substrates of the insulin receptor kinase and contains multiple tyrosine phosphorylation motifs that serve as docking sites for SH2 domains that mediate the metabolic and growth-promoting functions of insulin. 7 IRS-1 also contains over 30 potential serine/threonine phosphorylation sites. Phosphorylation of IRS-1 at serine/threonine inhibits insulin stimulation of tyrosine phosphorylation, resulting in inhibition of downstream insulin signaling molecules. 6 It has been shown that phosphorylation of IRS-1 at Ser612 (human Ser616) and Ser307 (human Ser312) inhibits the insulin/PI3K signaling pathway. 6 The phosphorylation of IRS-1 at Ser612 causes dissociation of the p85 subunit of PI3K, inhibiting downstream signaling, whereas the phosphorylation of IRS-1 on Ser307 results in its dissociation from the insulin receptor and triggers proteasome-dependent degradation. 6 Ang II induces serine phosphorylation at both sites and inhibits downstream signaling, including Akt phosphorylation, Glut-4 translocation to the sarcolemma, and NO production in endothelium.6,7 Studies in the TG(mREN2)27 rat model, which harbors the mouse Ren-2 renin gene and is a transgenic model with elevation of systemic and tissue Ang II levels, 50 have shown that these rats exhibit whole-body and skeletal muscle insulin resistance, associated with enhanced ROS production and defective activation of the IRS-1/PI3K signaling pathway.51,52 Furthermore, the antioxidant tempol improved insulin-mediated glucose uptake in the skeletal muscle of this animal model, 51 suggesting that Ang II impairs insulin sensitivity at least in part through generation of ROS.
In the vasculature, insulin stimulates two major signaling transduction cascades: PI3K and MAPK. 4 Insulin stimulation of NO production through activation of the PI3K pathway leads to vasodilation and increased blood flow, and subsequent augmentation of glucose disposal in skeletal muscle. 4 NO is an important vasoprotective molecule. In physiological conditions, constitutive stimulation of NO production by insulin may play an important role in the maintenance of vascular health as well as regulation of glucose metabolism. 22 Insulin also stimulates the MAPK pathway in the vascular endothelium, which mediates cellular growth and migration as well as production of prothrombotic and profibrotic factors. 35 The level of insulin necessary for stimulation of MAPK and PI3K may differ. 35 Fasting plasma insulin levels in a normal insulin-sensitive individual are usually in the low picomolar range (50–150 pM). At this range, insulin constitutively stimulates the PI3K pathway, which participates in the regulation of the metabolic effects of insulin and maintenance of vascular tone. In insulin-resistant states, such as obesity and diabetes, fasting insulin levels may reach the nanomolar range and are often associated with activation of RAAS. In addition, insulin-stimulation of the PI3K pathway is selectively impaired. 35 Overactivation of RAAS and hyperinsulinemia may synergistically stimulate the MAPK pathway, which exerts detrimental effects on the vascular wall by inducing endothelial dysfunction and promoting atherosclerosis. 4 Several clinical and experimental studies1,53–55 have shown that inhibition of RAAS by either ACE inhibitors, ARBs, or mineralocorticoid receptor (MR) antagonists improve insulin signaling and insulin sensitivity, indicating an important role of RAAS in impaired insulin signaling. However, several clinical studies have failed to demonstrate any beneficial effects of ARBs on insulin resistance.56–58 The reasons for this discrepancy may include differences in study design and/or study populations, and suggest the involvement of complex mechanisms underlying the interaction between RAAS and insulin resistance.
RAAS, insulin resistance and obesity
Obesity is a common cause of insulin resistance and one of the strongest risk factors for the development of T2DM as well as CV diseases. 59 Many studies suggest that obesity is associated with a systemic chronic inflammatory response characterized by altered proinflammatory cytokine production and activation of inflammatory signaling pathways in adipose tissue.47,60 It is increasingly recognized that adipose tissue does not only store energy, but acts as an active endocrine and paracrine organ that produces and releases a large number of cytokines and bioactive mediators, referred to as adipocytokines, that regulate insulin signaling. 61 The inflammatory cytokines or diabetogenic adipokines are mostly produced from pre-adipocytes (undifferentiated adipocytes), whereas differentiated adipocytes produce antidiabetic adipocytokines, such as adiponectin.62,63 The adipocytes express all components of RAS, including angiotensinogen, ACE, AT1, and AT2 receptors. 62 Experimental studies have provided evidence showing that Ang II inhibits adipogenic differentiation of human adipocytes via the AT1 receptor, resulting in increased secretion of inflammatory cytokines or diabetogenic adipokines, leading to inhibition of insulin signaling and insulin resistance. 64 It has been proposed that blockade of RAAS may promote the recruitment and differentiation of pre-adipocytes and increased formation of small insulin-sensitive adipocytes, thereby improving insulin sensitivity. 46 In the OLEFT rat, an animal model with T2DM, Lee et al. 46 showed that treatment with an ARB improved the differentiation of adipocytes and inhibited activation of the inflammatory process in adipose tissue, accompanied by an increase in adiponectin and a decrease in NFκB, PAI-1, and MCP-1.
RAAS, insulin resistance and salt-sensitive hypertension
Hypertension and T2DM are two powerful risk factors for development of CV disease. The salt sensitivity of blood pressure and insulin resistance have been identified as key factors underlying the relationship between hypertension and T2DM.1,65 Excess dietary salt and caloric intake, as commonly found in westernized diets, is linked not only to increased blood pressure, but also to impaired insulin sensitivity and glucose homeostasis. 1 A recent clinical study 66 has shown that the insulin resistance syndrome enhances the blood pressure response to sodium intake. Thus, a reduction in sodium intake could be an important component in reducing blood pressure in patients with multiple risk factors for the insulin resistance syndrome.
There is a strong clustering of markers of endothelial damage in individuals predisposed to salt-sensitive hypertension who concomitantly have insulin resistance and microalbuminuria. 67 Clinical studies have demonstrated that the salt sensitivity of blood pressure is an independent risk factor for increased CV morbidity and mortality in normotensive and hypertensive individuals. 67 Salt-sensitive hypertensive patients account for about 50% of hypertensives and have a higher incidence of left ventricular hypertrophy, endothelial dysfunction, hyperlipidemia, and microalbuminuria compared with salt-resistant hypertensive patients. 68 The Dahl salt-sensitive (DS) rat is an animal model of low-renin salt-sensitive hypertension in humans, characterized by CV disease and insulin resistance. Using this model, we and others have shown that salt-sensitive hypertension is a vascular diathesis characterized by impairment of endothelial NO-dependent vasorelaxation, upregulation of vascular tissue Ang II action, and ROS production, as well as susceptibility to CV and renal injury.69,70 Moreover, we have shown that the hypertensive DS rats manifest metabolic insulin resistance, determined by hyperinsulinemic euglycemic clamp, impaired endothelium-dependent relaxation to insulin (vascular insulin resistance), and impaired insulin signaling through the PI3K/Akt/NO pathway, associated with increased vascular and systemic inflammatory responses.9,71 Treatment with the ARB candesartan or the antioxidant tempol improved metabolic insulin sensitivity and insulin-mediated vasorelaxation (Figure 3) via insulin-mediated stimulation of NO production, suggesting that Ang II-mediated ROS overproduction interferes with insulin signaling pathways in both metabolic and CV tissues in this hypertensive animal model. 9
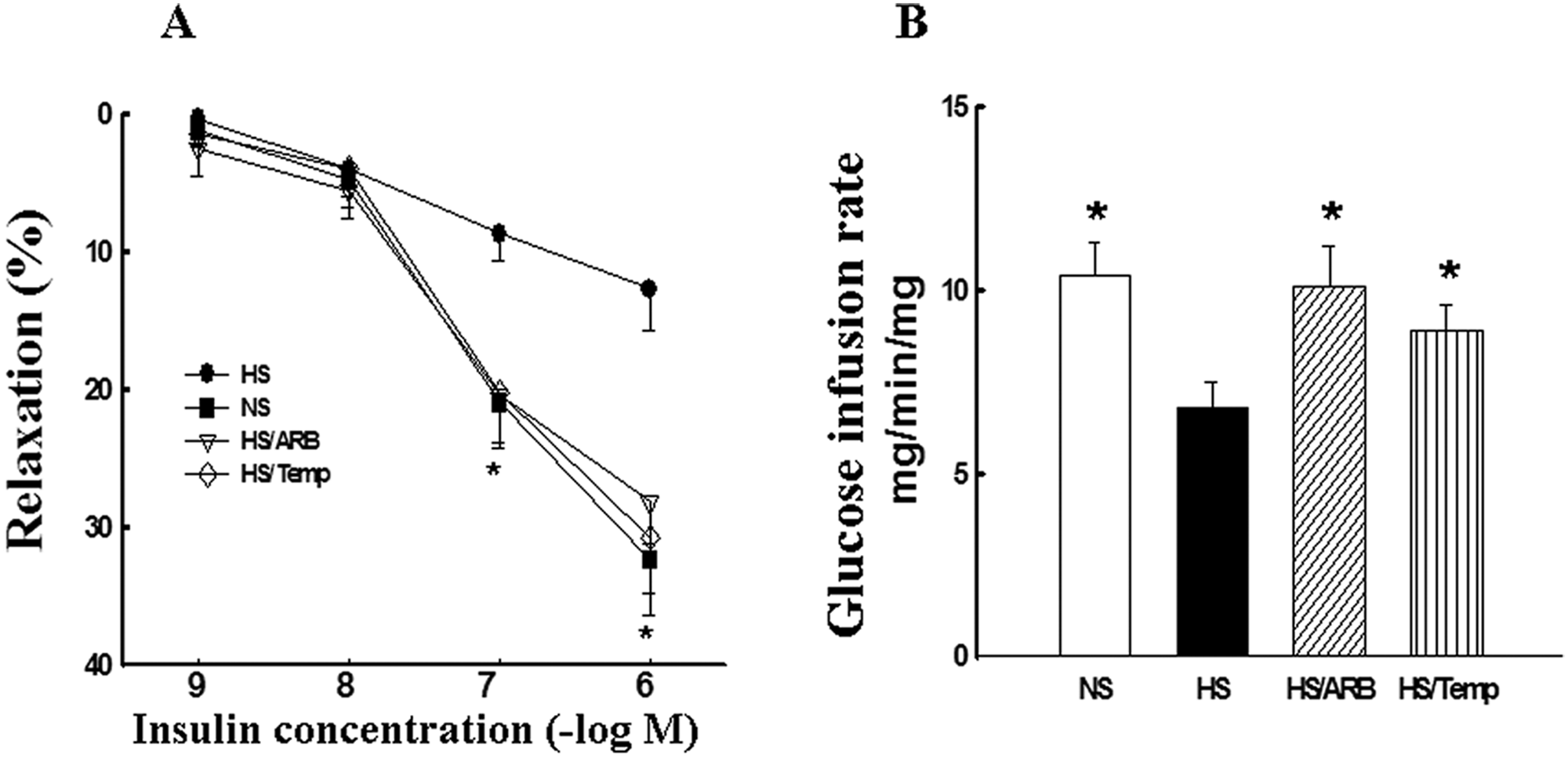
Effects of angiotensin type 1 receptor blocker (ARB) and antioxidant tempol on insulin-mediated vasorelaxation (A) and glucose disposal (B: glucose infusion rate) in Dahl salt-sensitive (DS) rats. DS rats were fed normal salt (0.5% NaCl, NS), high salt (4% NaCl, HS), HS plus ARB candesartan (10 mg/kg/day in drinking water, HS/ARB), or HS plus tempol (172 mg/l in the drinking water, HS/Temp) for 6 weeks. Endothelium-dependent relaxation to insulin in the aorta was determined using an organ chamber bath. Glucose infusion rate, an index of insulin sensitivity, was determined by hyperinsulinemic-euglycemic clamp. Insulin-induced vasorelaxation was significantly impaired in the aorta of hypertensive rats and was restored by either ARB or tempol treatment (A). The glucose infusion rate required to maintain plasma glucose at 5.5 mmol/l during the insulin-infusion period was also significantly reduced in hypertensive DS rats and significantly improved with ARB or tempol treatment (B), suggesting that Ang II and ROS is involved in impaired systemic and vascular insulin resistance. The data are expressed as mean ± SEM; *p < 0.05 vs HS; n = 6–7.
RAAS, insulin resistance and endothelial dysfunction
The endothelium represents a single layer of cells that line all the vessels in the body, including the conduit and resistance vessels, precapillary arteries, and capillaries. The vascular endothelium regulates local vasomotor tone, local homeostasis, and vascular wall proliferation processes via release/synthesis of numerous vasoactive factors referenced as endothelium-derived factors. Endothelium-derived NO is a well characterized relaxing factor that contributes to the overall regulation of arterial blood pressure by virtue of its ability to relax vascular smooth muscle. 72
Insulin resistance and endothelial dysfunction are two major components of the metabolic syndrome. 73 Endothelial dysfunction, characterized by reduced NO bioavailability, contributes to CV disease, including hypertension, atherosclerosis, and coronary artery disease, which are also characterized by insulin resistance. 12 In the context of the vascular system, insulin resistance manifests as impaired vasodilation, microvascular disease (i.e. retinopathy, neuropathy, and nephropathy), and enhanced vascular inflammation and atherosclerotic lesion formation. 4 Thus, impairment of insulin action in the vascular endothelium may be a link between hemodynamic and metabolic dysfunction in obesity and T2DM4. Indeed, obese individuals with insulin resistance and patients with T2DM have impaired insulin-stimulated activation of PI3K. In these patients, endothelium-dependent relaxation in response to insulin as well as acetylcholine is often blunted. 74
Accumulating clinical and experimental evidence indicates that activation of the RAS plays an important role in the development of endothelial dysfunction and insulin resistance in CV diseases and T2DM.46,75 Activation of RAS has been shown to be related to impaired insulin signaling and systemic insulin resistance in numerous tissues, including skeletal muscle and liver, as well as cardiac, vascular, and renal tissues.48,49 Studies in humans have demonstrated a significant association between insulin resistance and hypertension, which might be mediated by RAAS activation and oxidative stress.67,76 It is well established that activation of RAAS contributes to endothelial dysfunction through increased nicotinamide adenine dinucleotide phosphate (NADPH) oxidase-derived ROS mechanisms in hypertension and other CV diseases. 77 Hypertension and T2DM are also associated with a decreased number and impaired function of endothelial progenitor cells (circulating bone marrow-derived stem cells that play an important role in vascular wall endothelial repair). 78 Inhibition of the RAAS increases the number and function of endothelial progenitor cells in patients with stable coronary artery diseases as well as in subjects with T2DM.78,79 Indeed, clinical and experimental studies have demonstrated that ACE inhibitors, ARBs, MR blockers and antioxidants improve endothelium-dependent vasodilation, arterial structure, as well as insulin sensitivity in diabetic and hypertensive patients and animals, highlighting an important role of RAAS and ROS in the induction of vascular diseases and impaired insulin sensitivity.49,77,80
RAAS blockade in the clinical setting
The effects of RAAS blockade with ACE inhibitors and ARBs have been studied extensively in patients with hypertension, congestive heart failure, coronary artery disease, and chronic kidney disease.11,81 RAAS inhibition has been shown to improve insulin resistance and glucose intolerance in non-diabetic patients with CV disease or CV risk factors, as well as improve CV and renal outcomes in diabetic patients.56,80,82,83 In addition, results from several large randomized clinical trials, including VALUE, 84 NAVIGATOR, 85 ALLHAT, 86 HOPE, 87 CAPPP, 88 CHARM, 89 SOLVD 90 and LIFE, 91 suggest that ACE inhibitors and ARBs reduce the incidence of new-onset T2DM (NOD) in high-risk patients with CV disease.
The VALUE trial was designed to compare CV outcomes in treatment regimens based on the ARB valsartan and the calcium channel blocker amlodipine in a population of essential hypertensive patients recruited according to a specific predefined age- and risk factor-dependent algorithm. A total of 15,245 high-risk patients were followed for an average of 4.2 years. The results showed that there was no difference between the two drug regimens in the primary composite cardiac endpoint rate or in all-cause mortality. 84 However, the relative risk of NOD, a pre-specified secondary end point, was 23% lower in the group receiving valsartan than in the amlodipine group.55,84,92
The findings from the VALUE trial showing that RAAS inhibition prevents the development of NOD have been supported by a number of other clinical studies.85,86 The NAVIGATOR trial demonstrated that treatment with the ARB valsartan during an average of 5 years, in addition to lifestyle modifications, reduced the progression to T2DM in a large group of patients with impaired glucose tolerance and established CV disease.85,93 A meta-analysis of 13 randomized trials was done to determine if ACE inhibitors and/or ARBs prevent the development of T2DM. 94 These trials enrolled 92,408 patients without T2DM, of whom 41,950 were randomized to treatment with an ACE inhibitor or ARB, while the remainder received active control (calcium channel blockers (CCBs), thiazide diuretics, or β-blockers) or placebo. RAAS inhibition resulted in a 27% reduction in the relative risk of NOD in nine of these trials with hypertensive patients, and a 33% reduction of NOD in four of the trials with patients with vascular disease or left ventricular dysfunction. On the other hand, in the DREAM (Diabetes Reduction Assessment with Ramipril and Rosiglitazone Medication) trial, 95 the ACE inhibitor ramipril was not associated with a significant reduction in development of NOD compared with placebo. However, ramipril significantly increased regression to normoglycemia relative to placebo administration, suggesting a beneficial effect of RAAS blockade on glucose homeostasis.
Patients with T2DM have a high risk of development of CV complications. Several recent large CV outcome trials conducted among patients with T2DM have demonstrated that intensive glycemic control with traditional anti-diabetic agents failed to lower the risk of incident CV diseases.96–99 The lack of CV benefit of traditional anti-diabetic agents may be due to their inability to decrease CV inflammation, 100 which is mechanistically linked to the development of CV disease and the metabolic syndrome. ACE inhibition and ARBs exert modest effects on glycemia but significant inhibitory effects on systemic and/or local (renal, CV, adipose, and skeletal muscle) inflammation and oxidative stress.83,101 Randomized trials have established the CV benefits of ACE inhibitors and ARBs in patients with T2DM. Therefore, ACE inhibitors and ARBs are currently recommended for the prevention and treatment of CV diseases in patients with T2DM.
Activation of the AT1 receptor by Ang II can trigger some of its actions through interaction with and activation of aldosterone-mineralocorticoid receptor signaling. 1 Aldosterone has been shown to upregulate the expression of ACE in cultured neonatal rat cardiomyocytes. 102 Aldosterone excess results in increased ROS production in CV tissues through activation of the membrane-bound NADPH oxidase. It has been shown that aldosterone through stimulating ROS generation inhibits insulin signaling molecules IRS-1 or IRS-2 in 3T3-L1 adipocytes and vascular smooth muscle cells,103,104 as well as glucose-mediated insulin release in murine islets. 105 From a clinical perspective, excess aldosterone is linked to impaired glucose tolerance and T2DM. Current data indicates that up to one-half of patients with primary hyperaldosteronism have impaired glucose tolerance.54,76 Several clinical studies have demonstrated a significant association of aldosterone plasma levels with insulin resistance and there is some evidence that plasma aldosterone levels may be predictive of the development of insulin resistance.106,107 Moreover, insulin resistance is restored by surgical intervention or MR antagonists in primary hyperaldosteronism. 108 Clinical studies in patients with primary hyperaldosteronism have also revealed that excess aldosterone is related to increased cardiac mass, fibrosis, and cardiovascular tissue remodeling relative to those in individuals with essential hypertension. 76 Inhibition of aldosterone by aldosterone synthase inhibitors or MR antagonists improves CV outcomes in patients with CV disease or CV risk factors.109–111
A reactive increase in the activity of renin has been observed when either ACE inhibitors or ARBs are used for long periods of time. 112 Renin is a rate-limiting enzyme in RAAS activation. It now appears that renin exerts additional actions through a renin receptor, distinct from its actions that lead to the production of Ang II and aldosterone. 113 It is anticipated that the ability to inhibit renin would improve blood pressure control and might modify other actions and interactions of this important vasoactive system.114,115 Pharmacological inhibition of the renin receptor-dependent system has shown beneficial effects in diabetic nephropathy and retinopathy in diabetic patients and animal models.113,116,117 Clinical studies have shown that direct renin inhibition with aliskiren is effective in promoting left ventricle (LV) mass regression in hypertensive patients with LV hypertrophy. 118 Inhibition of renin with aliskiren attenuated systemic insulin resistance, oxidative stress, and pancreatic remodeling and improved skeletal muscle glucose insulin transport in the transgenic renin 2 rats, obese Zucker rats,119,120 suggesting a role of renin in the development of insulin resistance. However, whether inhibition of renin improves insulin resistance in the clinical setting remains to be investigated.
Experimental and clinical studies have established the beneficial effects of RAAS inhibition in CV and metabolic diseases. However, the mechanisms underlying the improvement of insulin resistance and CV injury by RAAS blockade are complex and not fully elucidated. Both Ang II and aldosterone stimulate release of inflammatory cytokines and ROS production, leading to inhibition of insulin signaling and endothelial dysfunction. The mechanisms that have been proposed include stimulation of intracellular insulin signaling in insulin-sensitive and CV tissues,13,75 improvement of microvascular endothelial function leading to increased blood flow and glucose delivery and disposal in skeletal muscle, 12 improvement of pancreatic β-cell function resulting in enhanced insulin secretion in response to glucose stimulation, 46 and modulation of adipose tissue hormonal responses 121 (Figure 4).
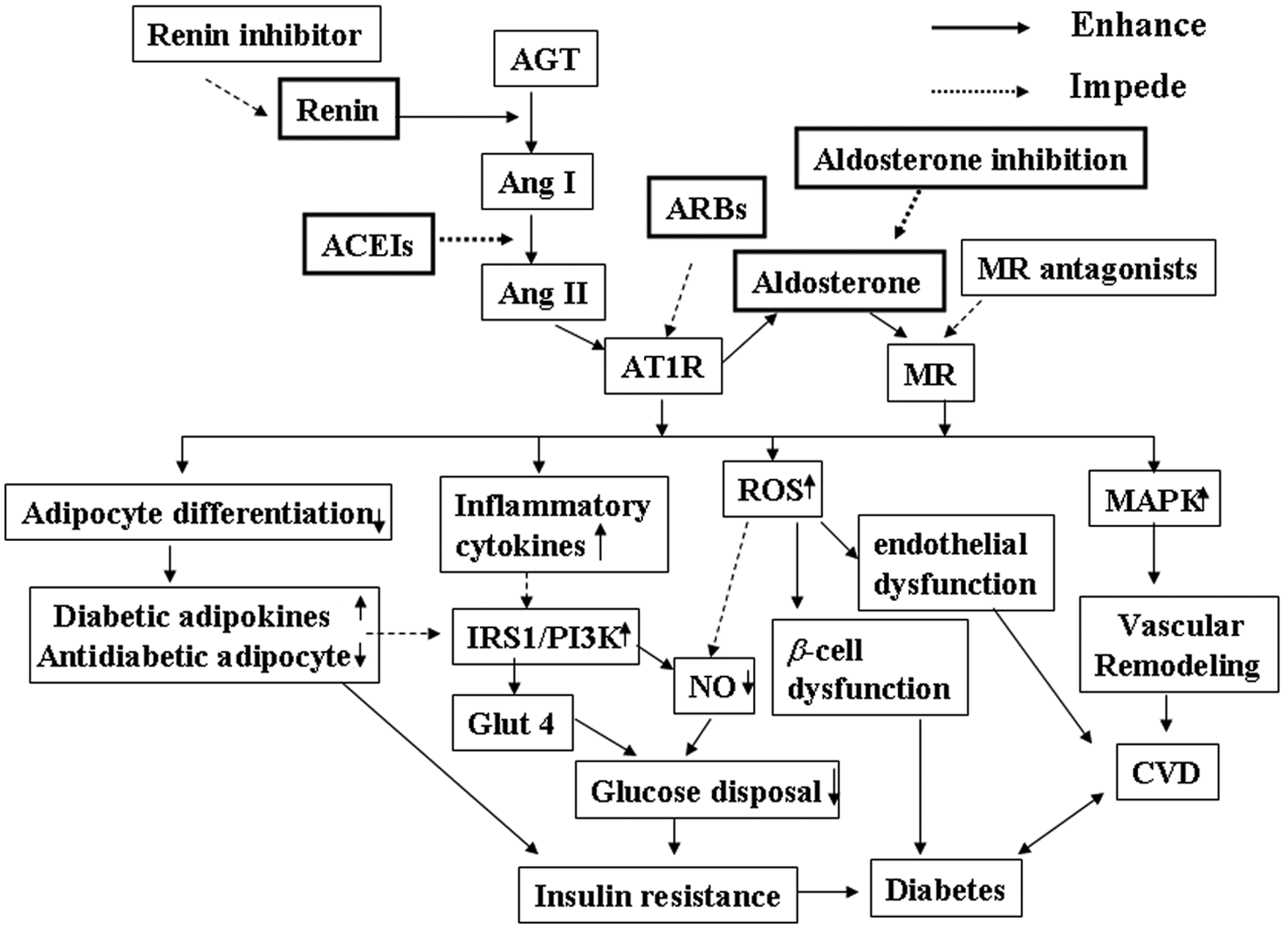
Schemata of potential mechanisms underlying the improvement of insulin resistance and prevention of diabetes development by inhibition of the renin–angiotensin–aldosterone system (RAAS). In the RAAS system, renin cleaves a leucine–valine bond in the angiotensinogen (AGT) molecule to release angiotensin I (Ang I), which is cleaved to Ang II by angiotensin-converting enzyme (ACE). Ang II induces aldosterone release and other biological effects though activation of the AT1 receptor. Activation of Ang II and aldosterone produces the following major biological effects: (1) inhibition of insulin activation of the insulin receptor substrate-1 (IRS-1)/phosphatidylinositol 3-kinase (PI3K) signaling pathway through increased inflammatory cytokine release in insulin-sensitive tissues and cardiovascular (CV) system; (2) inhibition of pre-adipocyte differentiation into mature adipocytes, leading to an increase in secretion of diabetic adipokines and a decrease in antidiabetic adipokines; (3) increased reactive oxygen species (ROS) production, resulting in islet cell structural damage, β-cell dysfunction and endothelial cell dysfunction; and (4) activation of the mitogen-activated protein kinase (MAPK) pathway, leading to vascular remodeling and promotion of CV disease (CVD). All of these biological effects cause impairment of glucose metabolism and insulin resistance, thus contributing to the development of diabetes. Inhibition of RAAS, including renin inhibitors, ACE inhibitors (ACEI), angiotensin II type 1 receptor blockers (ARBs), aldosterone synthase inhibitors and mineralocorticoid receptor (MR) antagonists, prevents these detrimental effects of Ang II and/or aldosterone on glucose metabolism, therefore improving insulin sensitivity, and potentially reducing the development of diabetes and CV complications.
Conclusion
Approximately 25% of adults in the USA have been diagnosed with the metabolic syndrome. Individuals with the metabolic syndrome have an increased risk of coronary artery disease, which translates into a 2.6-fold increase in CV mortality. Accumulating experimental evidence indicates that activation of the RAAS is critical for the development of insulin resistance, glucose intolerance, T2DM, hypertension, and other CV diseases.5,9,49,71 Further research is necessary to identify patients at risk for the development of T2DM and other CV complications and those who would most benefit from RAAS blockade.
Footnotes
Funding
This work was supported by an American Heart Association National Scientist Development Award and Florida JEK Biomedical Research grant to Ming-Sheng Zhou.
Conflict of interest
None declared.