
Abstract
Intermittent claudication (IC) is the predominant symptom of peripheral artery disease (PAD), and is associated with reduced exercise capacity. The pathophysiology of IC is related to reduced blood flow and impaired skeletal muscle oxidative metabolism; however, the efficacy of metabolic therapies is not well established. We evaluated the effect of cilostazol plus l-carnitine versus cilostazol alone on exercise performance, quality of life (QOL), and safety. In a double-blind, placebo-controlled trial, PAD patients with stable IC were randomized to either l-carnitine 1 g or matching placebo twice-daily, on a background of cilostazol. Treadmill and QOL assessments were performed at baseline, 90, and 180 days. The primary endpoint was the difference between groups in the natural-log-transformed (ln) ratio in peak walking time (PWT) between baseline and 180 days. The combination of cilostazol and l-carnitine was well tolerated. In the modified intent-to-treat population (n = 145), the mean ln ratio in PWT was 0.241 for cilostazol/l-carnitine versus 0.134 for cilostazol/placebo (p = 0.076), corresponding to mean increases of 1.99 and 1.36 minutes, respectively. In the per-protocol population (n = 120), the mean ln ratio in PWT was 0.267 for cilostazol/l-carnitine versus 0.145 for cilostazol/placebo (p = 0.048). QOL measures were also improved in the cilostazol/l-carnitine group. These findings support larger trials of l-carnitine in combination with cilostazol in the treatment of IC.
Introduction
Peripheral artery disease (PAD) is a manifestation of systemic atherosclerosis and a strong predictor of cardiovascular morbidity and mortality. Symptoms can range from asymptomatic to atypical leg pain to intermittent claudication (IC), characterized by cramping, aching, or fatigue in the calf muscles provoked by activity and relieved by rest. The pathophysiology of IC in PAD is thought to relate to inadequacy in the blood flow to supply oxygen and the key nutrients required to meet the metabolic demands of exercising skeletal muscle, leading to impaired oxidative metabolism at the end-organ (i.e. skeletal muscle) level. 1 Alterations in carnitine metabolism, resulting in muscle accumulation of short-chain acylcarnitines, have been described in patients with IC due to PAD, 1 and have been proposed as an additional mechanism to explain exercise limitation in these patients.
The only FDA-approved medical therapies for IC are pentoxifylline and cilostazol, an oral phosphodiesterase (PDE)-III inhibitor. Cilostazol has been evaluated in meta-analyses2–4 of randomized controlled clinical trials (RCTs)5–10 that have demonstrated a significant improvement in maximal walking distance and pain-free walking distance or time on graded exercise treadmill testing, as well as quality of life (QOL), relative to placebo. In addition, metabolic interventions aimed at treating the abnormal skeletal muscle metabolism, such as l-carnitine and propionyl-l-carnitine supplementation, have been proposed for the treatment of IC in PAD. Carnitine facilitates long-chain fatty acid transfer from the cytosol of the cell to the mitochondrion, where beta-oxidation ensues and, ultimately, adenosine triphosphate (ATP) is generated. In addition, carnitine acts as an intracellular buffer for acyl coenzyme A (CoA), freeing acyl CoA for subsequent ATP production. While l-carnitine was evaluated several decades ago, a related compound, propionyl-l-carnitine, has been more extensively studied in PAD with several positive Phase 3 clinical trials.11–15 Based upon evidence of the efficacy and safety of both cilostazol2–10,16,17 and carnitine11–15,18–26 in IC trials, and in consideration of cilostazol as the established mainstay of pharmacological therapy in this disease, the present trial sought to investigate the effect of cilostazol plus l-carnitine versus cilostazol alone on treadmill performance in IC. Secondary objectives were to evaluate QOL measures and safety indices with the drug combination.
Methods
Design
This was a randomized, multicenter, parallel-group, double-blind, placebo-controlled trial designed to quantify the effects of l-carnitine on a background of cilostazol compared to the established therapy of cilostazol alone. Qualifying subjects (see ‘Subjects’) began a cilostazol run-in period which consisted of twice-daily administration of cilostazol 50 mg for approximately 2 weeks, followed by dose escalation to 100 mg cilostazol twice-daily if tolerated. Subjects intolerant of cilostazol 100 mg were allowed to continue at 50 mg with the approval of the medical monitor.
After completion of the cilostazol run-in period, subjects continuing to meet inclusion and exclusion criteria were randomized via a centrally generated computer scheme to receive either l-carnitine 1 g twice-daily or placebo-matching l-carnitine, for 180 days. A sub-study was conducted to confirm the presence of an adequate pharmacokinetic (PK) peak response to l-carnitine administration, such that outcomes in the main study could be interpreted in the context of knowledge that l-carnitine-treated subjects demonstrated an increase in plasma l-carnitine levels. Safety and efficacy assessments were conducted at day 90 and day 180, with intervening telephone calls to assess compliance and tolerability. Study medication compliance was assessed by pill counts at the baseline (post-run-in period), day 90, and day 180 visits. With regard to endpoint quality assurance, all exercise tests were centrally adjudicated by an independent committee for assessment of protocol adherence. Blinding was maintained at the level of both the study staff (investigator, study coordinator) and the subject.
The trial was reviewed and approved by the local Institutional Review Boards (IRB) at all participant institutions or by a central IRB, as appropriate. Written informed subject consent was obtained prior to study participation in all cases. The trial was conducted in accordance with the US Code of Federal Regulations guidance pertaining to clinical studies (21 CFR parts 11, 50, 54, 56 and 312), the principles of the Declaration of Helsinki, and the guidelines of the International Conference on Harmonisation (ICH), and was registered on www.clinicaltrials.gov prior to trial initiation (ClinicalTrials.gov Identifier: NCT00822172).
Subjects
Subject inclusion criteria were: (1) ≥ 40 years old with IC due to PAD; (2) ankle–brachial index (ABI) < 0.90 in at least one extremity (if ABI was ≥ 0.90 to ≤ 1.0, then a reduction of at least 20% in ABI in at least one extremity was required when measured within 1 minute after claudication-limiting treadmill testing; if the subject had non-compressible arteries then a toe–brachial index (TBI) < 0.70 was required in at least one extremity); (3) symptoms of IC must have been stable for at least 3 months prior to first screening visit; (4) peak walking time (PWT) of ≥ 1 to ≤ 12 minutes on a Gardner protocol at the second screening visit; and (5) during the cilostazol tolerance (run-in) phase of the screening period, the subject demonstrated at least 70% compliance with cilostazol and was willing to continue treatment. ABI was defined as the ratio between the higher of the two pedal systolic blood pressures (dorsalis pedis and posterior tibialis) and the higher of the two systolic brachial pressures. TBI was defined as the ratio between the systolic pressure of the great toe and the higher of the two arm systolic pressures. Main exclusion criteria were: (1) critical limb ischemia; (2) leg amputation; (3) history of congestive heart failure; (4) active malignancy; (5) anticipated survival < 2 years; (6) transient ischemic attack or deep venous thrombosis in past 3 months; (7) stroke, coronary revascularization, or peripheral revascularization in the past 6 months; (8) resting blood pressure > 180/100 mmHg at screening; (9) currently taking, or unwilling to undergo 6-week wash-out from l-carnitine, cilostazol, or pentoxifylline; (10) currently taking, or unwilling/unable to discontinue ketoconazole, itraconazole, or erythromycin; (11) anticipated change in status with regard to smoking or physical activity; (12) current/anticipated pregnancy or breastfeeding during the study; and (13) any of the following laboratory abnormalities at screening: white blood cell count < 3.0 × 103/ml or > 15 × 103/ml; hemoglobin < 10 g/dl; platelet count < 100 × 103/ml; serum creatinine > 2.5 mg/dl; hemoglobin A1c > 10; alanine aminotransferase, aspartate aminotransferase, or total bilirubin > three times the upper limit of normal.
Endpoint evaluation
The primary efficacy endpoint was the change from baseline in PWT at day 180. PWT was defined as the maximum time walked on a graded treadmill until claudication symptoms forced the cessation of exercise. Secondary endpoints consisted of change from baseline in PWT at day 90; change from baseline in claudication onset time (COT), defined as time until the onset of claudication symptoms, at day 90 and day 180; and QOL at day 90 and day 180. Graded treadmill testing consisted of the Modified Gardner Treadmill Protocol, in which the test began at 2 mph (3.2 km/h), 0% grade and increased by 2% grade every 2 minutes, to a maximum of 18% grade. Treadmill testing was initiated following a minimum rest period of 10 minutes (or 30 minutes if the patient experienced claudication symptoms when walking to the testing facility); subjects were asked to refrain from physical activity prior to treadmill assessments on the day of study visits. The acceptability of treadmill tests was determined by a blinded endpoint quality assurance committee.
QOL was measured first by the Walking Impairment Questionnaire (WIQ) and next by SF-36v2®, as previously described.27,28 Both instruments were conducted prior to treadmill testing. Safety assessments (see also Figure 1) included vital signs, routine laboratory testing, ECG and adverse events (AEs).
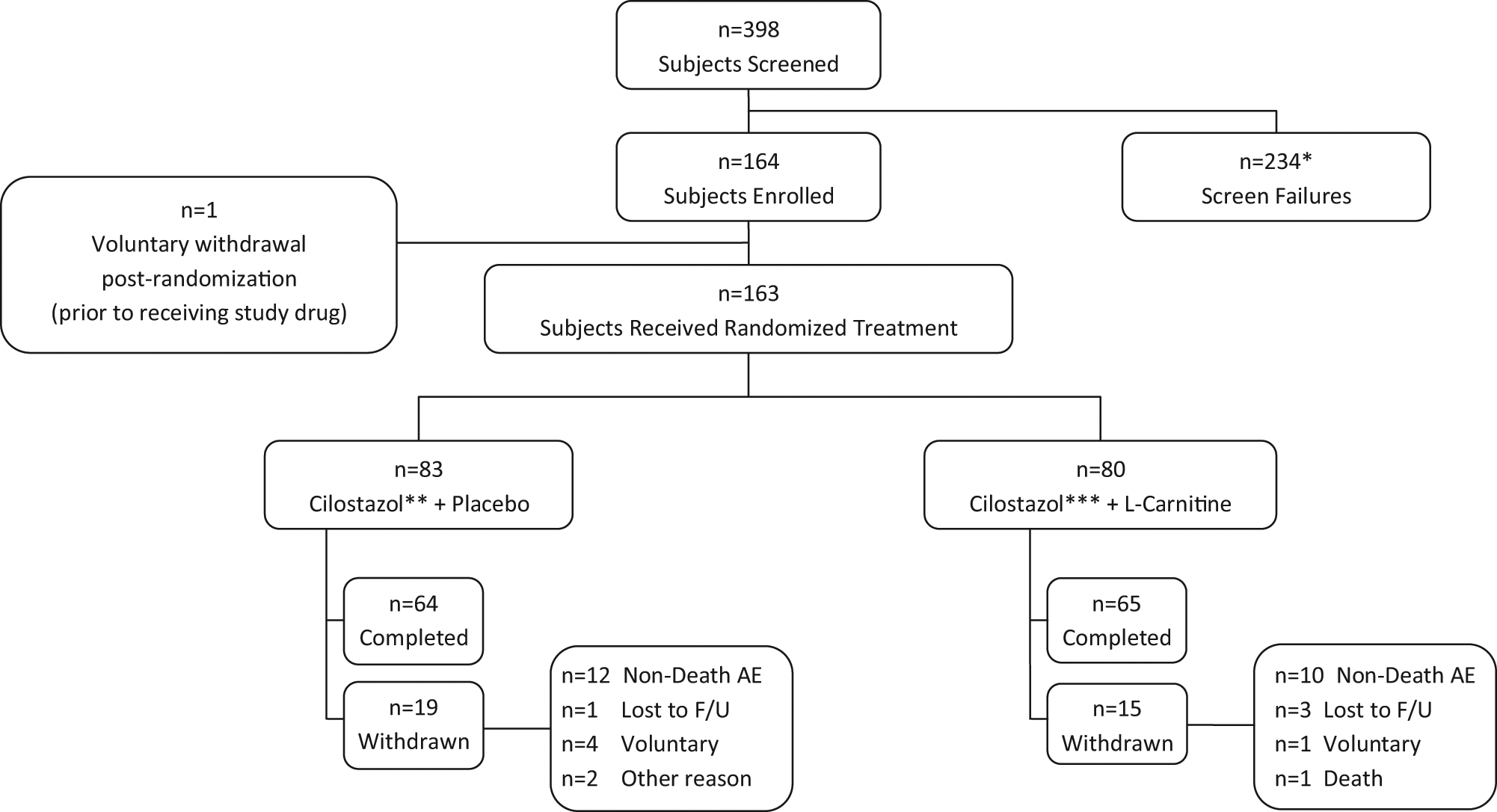
Subject disposition. (*Includes subjects who withdrew during the run-in period; **includes five subjects who remained on 50 mg cilostazol twice-daily due to suboptimal tolerability to 100 mg twice-daily during the run-in period; ***includes two subjects who remained on 50 mg cilostazol twice-daily due to suboptimal tolerability to 100 mg twice-daily during the run-in period.)
Statistical analysis
The primary efficacy analysis consisted of a comparison by Student’s t-test of the mean value of log-transformed (natural logarithm [ln]) ratios of PWT at day 180 to PWT at baseline for subjects in the cilostazol/l-carnitine group versus the cilostazol/placebo group, using the modified intention-to-treat (mITT) population. The mITT population was defined as all randomized subjects who received at least one dose of study medication and completed at least one post-randomization treadmill test. For each subject, a ratio was calculated of PWT at day 180 to PWT at baseline. The ln of this ratio was then taken in order to normalize the distribution of these data. The mean of the ln ratios was then reported by treatment group. To provide clinical context, arithmetic mean percent changes in PWT from baseline to day 180 were also reported by treatment group, using individual (per-patient) percent changes calculated as follows: percent change in PWT = [PWT (180 days) – PWT (baseline) / PWT (baseline)*100]. Missing day 180 data were replaced with the last post-randomization observation carried forward (either from the day 90 treadmill or an early termination visit treadmill).
A sample size of 83 subjects per arm provided at least 80% power to detect a statistically significant difference in mean log ratios (day 180/baseline) between the cilostazol/l-carnitine group and the cilostazol/placebo group, under the following assumptions: (1) cilostazol alone would result in a 40% increase in PWT (0.336 on a log scale); (2) cilostazol combined with l-carnitine treatment would result in a 60% increase in PWT (0.470 on a log scale); (3) the pooled standard deviation would be 0.38 on a log scale; and (4) the drop-out rate would be 15%. With these assumptions, 83 patients per group would provide 54% power in testing the primary hypothesis. If the increase in PWT was 68% then the trial would have 80% power.
Secondary efficacy analyses included: a repeat of the primary analysis in the per-protocol population (wherein the last observation carried forward must have been at least 150 days post-randomization); a repeat of the primary analysis in both the mITT and per-protocol populations, using the treadmill at the screening visit (i.e. prior to the cilostazol run-in period) as the ‘baseline’ PWT value; and mixed model analyses of COT, WIQ and SF-36v2® components in both the mITT and per-protocol populations. The per-protocol population was defined as all randomized subjects who were without major protocol deviations and who were at least 75% compliant with assigned treatment (calculated as the number of administered doses divided by the number of prescribed doses for the entire study period, multiplied by 100 to convert to percent). The walking distance summary measure on the WIQ was a weighted mean of the following distance components (i.e. weighted according to distance): indoors (20), 50, 150, 300, 600, 900, and 1500 feet (6.1, 15.2, 45.7, 91.4, 182.9, 274.3, and 457.2 meters). For the mixed model analyses: change from baseline was the response variable; treatment, time point, and treatment-by-time point interaction were fixed categorical effects; site and subject were random effects; and baseline (day 0) was a covariate. Because testing for a treatment-by-time interaction was not statistically significant, the mixed effects models were employed in the absence of this interaction, and were hence applied across the day 90 and day 180 time points.
Safety analyses were descriptive, summarizing the incidence, relatedness, severity and type of AEs and treatment-emergent changes in safety criteria (laboratory parameters, vital signs and ECG changes) by treatment group. The safety population consisted of all randomized subjects who received at least one dose of study medication.
The statistical analysis plan was completed and the database was locked prior to all analyses. For all inferential statistics, tests were two-sided and alpha was set at 0.05. Statistical analyses used SAS version 9.1 (SAS Institute, Cary, NC, USA).
Results
Subject disposition, demographics and compliance
A total of 164 subjects were enrolled in the trial; 145 met criteria for the mITT population and 120 for the per-protocol population. Subject disposition is shown in the Figure 1 flow diagram. Demographics are summarized by treatment group and overall in Table 1 for the mITT population. There were no statistically significant differences in baseline characteristics between treatment groups, and no differences were noted in demographic characteristics between the mITT and per-protocol populations. Mean (SD) compliance with randomized study medication was similar between treatment groups, at 94% (10%) in the cilostazol/l-carnitine group and 94% (8%) in the cilostazol/placebo group. Regarding tolerability of the combination of cilostazol with l-carnitine, 90% of patients in this treatment group demonstrated 75% or greater compliance with study drug.
Demographics, by treatment group, in the modified intention-to-treat (mITT) population
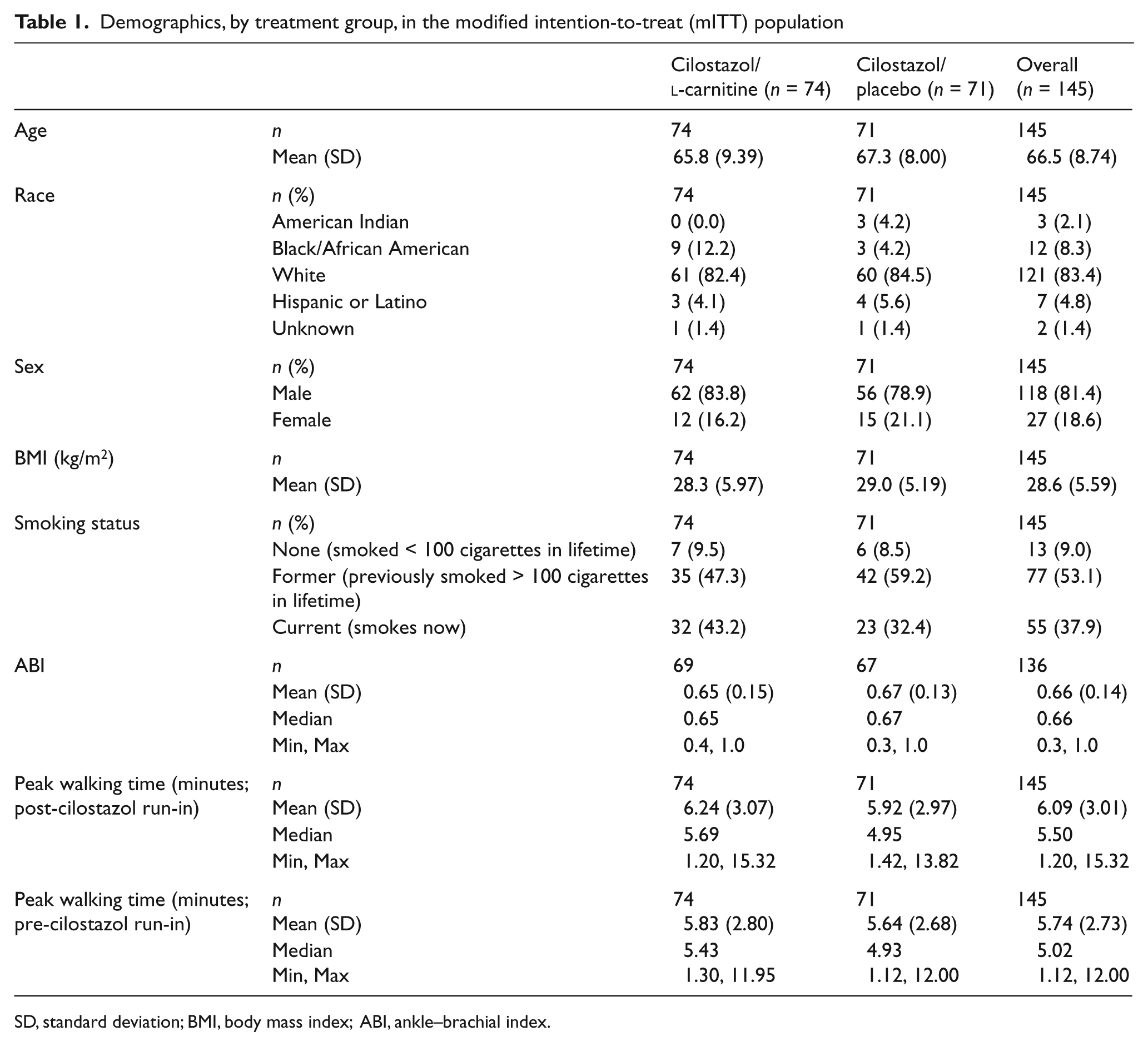
SD, standard deviation; BMI, body mass index; ABI, ankle–brachial index.
Primary efficacy analysis
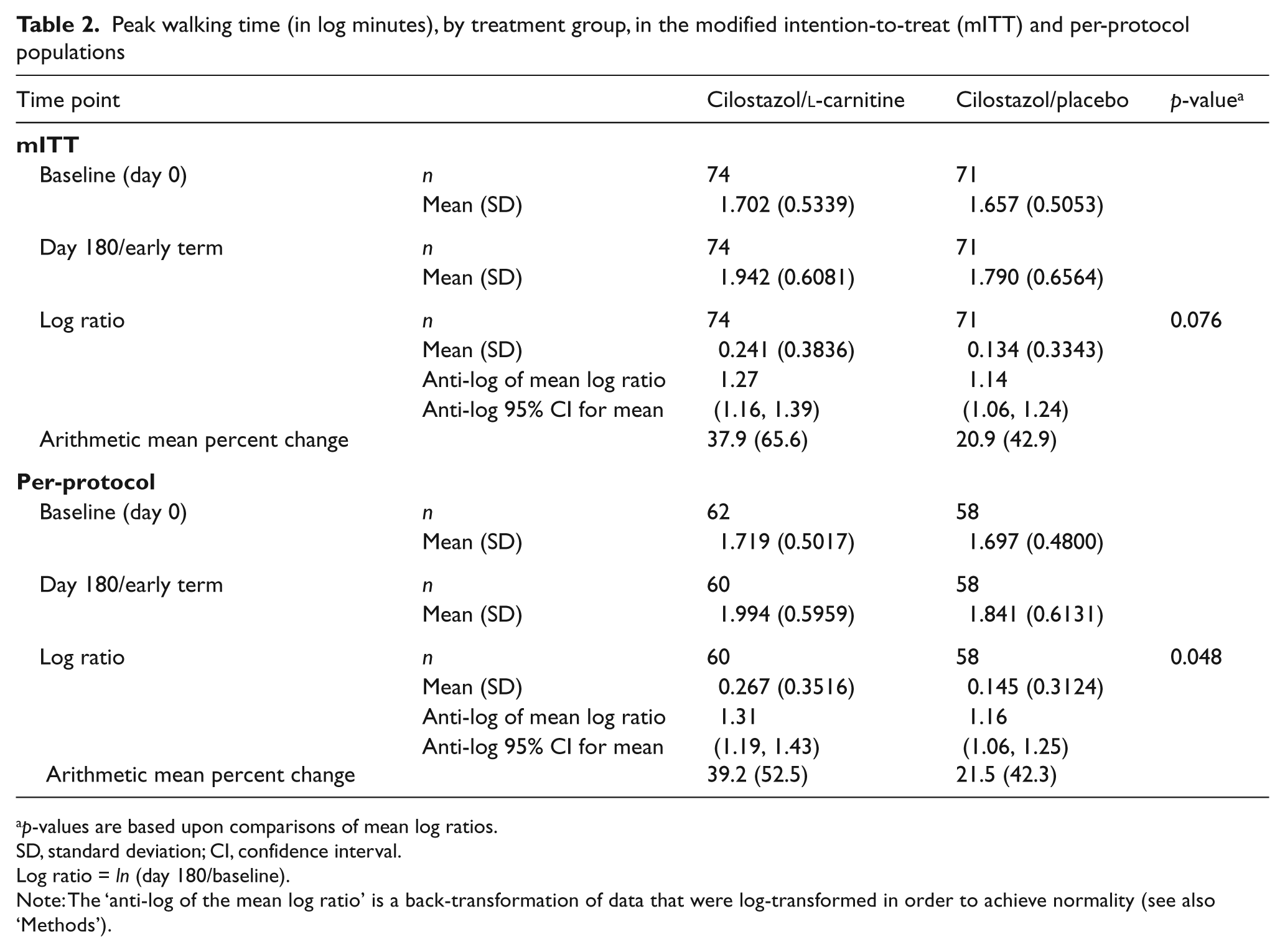
As shown in Table 2 for the mITT population, the mean ln ratio (day 180 versus baseline) in PWT was 0.241 for the cilostazol/l-carnitine group versus 0.134 for the cilostazol/placebo group (p = 0.076). This represented an arithmetic mean increase in PWT of 1.99 minutes (or 37.9%) from baseline to day 180 for cilostazol/l-carnitine, as compared with 1.36 minutes (or 20.9%) for cilostazol/placebo.
Peak walking time (in log minutes), by treatment group, in the modified intention-to-treat (mITT) and per-protocol populations
p-values are based upon comparisons of mean log ratios.
SD, standard deviation; CI, confidence interval.
Log ratio = ln (day 180/baseline).
Note: The ‘anti-log of the mean log ratio’ is a back-transformation of data that were log-transformed in order to achieve normality (see also ‘Methods’).
Secondary efficacy analyses
As shown in Table 2 for the per-protocol population, the mean ln ratio in PWT was significantly increased in the cilostazol/l-carnitine group versus the cilostazol/placebo group (0.267 vs 0.145, respectively; p = 0.048). This represented an arithmetic mean increase in PWT of 39.2% from baseline to day 180 for cilostazol/l-carnitine, as compared to 21.5% for cilostazol/placebo. Day 90 findings in the mITT and per-protocol populations showed no statistically significant differences between treatment groups.
When the second baseline visit (i.e. prior to the cilostazol run-in period) was used as ‘baseline’ in lieu of the day 0 visit, the mean ln ratios (day 180 versus the second screening visit) in PWT for cilostazol/l-carnitine versus cilostazol/placebo were 0.242 versus 0.198 (p = 0.445) in the mITT population and 0.349 versus 0.206 (p = 0.034) in the per-protocol population. Corresponding arithmetic mean increases in PWT for the mITT and per-protocol populations were 45.2% versus 31.4% and 51.1% versus 31.5%, respectively.
The mean change from baseline in COT in the mITT population across the day 90 and day 180/early termination time points, combined, was significantly increased in the cilostazol/l-carnitine group when compared to the cilostazol/placebo group (p = 0.018). With regard to QOL measures (Tables 3A and 3B), in the mITT population, the mean increase from baseline in walking distance on the WIQ across the day 90 and day 180/early termination time points, combined, for the cilostazol/l-carnitine group was nearly twice that of the cilostazol/placebo group, although this difference was not statistically significant (13.20 vs 6.57; p = 0.331). In the cilostazol/l-carnitine group, the mean increase in physical functioning on the SF-36v2® was also nearly double that of the cilostazol/placebo group [6.77 (16.379) vs 3.73 (17.566), respectively; p = 0.066].
Mixed model analysis a of walking distance in the Walking Impairment Questionnaire, by treatment group, in the modified intention-to-treat (mITT) and per-protocol populations
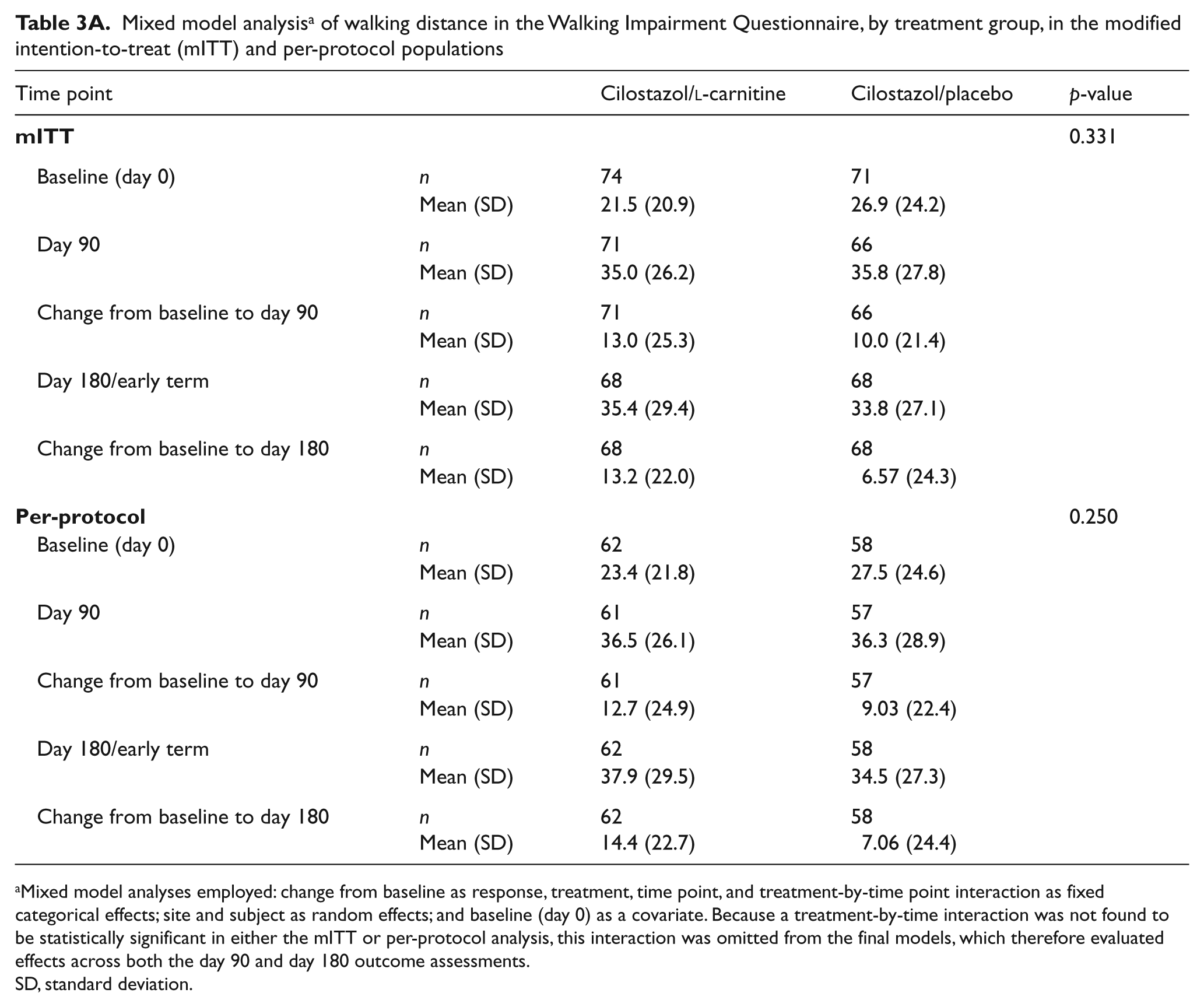
Mixed model analyses employed: change from baseline as response, treatment, time point, and treatment-by-time point interaction as fixed categorical effects; site and subject as random effects; and baseline (day 0) as a covariate. Because a treatment-by-time interaction was not found to be statistically significant in either the mITT or per-protocol analysis, this interaction was omitted from the final models, which therefore evaluated effects across both the day 90 and day 180 outcome assessments.
SD, standard deviation.
Mixed model analysis a of the physical functioning score on the SF-36, by treatment group, in the modified intention-to-treat (mITT) and per-protocol populations
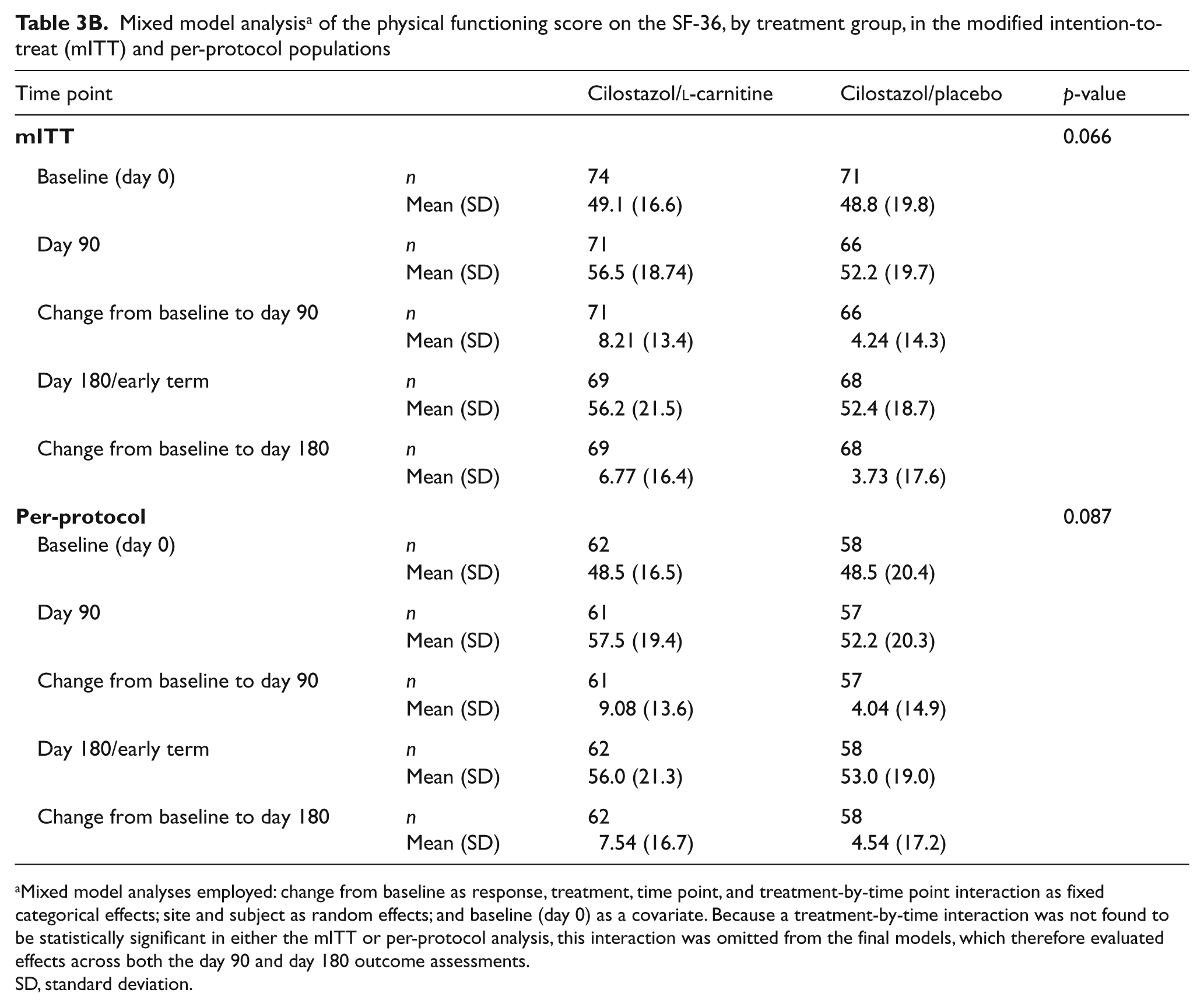
Mixed model analyses employed: change from baseline as response, treatment, time point, and treatment-by-time point interaction as fixed categorical effects; site and subject as random effects; and baseline (day 0) as a covariate. Because a treatment-by-time interaction was not found to be statistically significant in either the mITT or per-protocol analysis, this interaction was omitted from the final models, which therefore evaluated effects across both the day 90 and day 180 outcome assessments.
SD, standard deviation.
Safety analyses
Most common AEs and all serious AEs (SAEs) are summarized in Tables 4 and 5, respectively. Diarrhea was the most frequently observed AE, occurring in 11% of subjects in the cilostazol/l-carnitine group and in 7% in the cilostazol/placebo group. AEs characterized as either related or possibly related to study medication were observed in 11% of subjects receiving cilostazol/l-carnitine and 17% receiving cilostazol/placebo. With regard to SAEs, frequencies were similar between treatment groups. A total of seven cardiovascular SAEs were noted in the cilostazol/placebo group and five in the cilostazol/l-carnitine group. There was one death, consisting of fatal myocardial infarction in a patient randomized to cilostazol/l-carnitine with remote previous myocardial infarction, which was deemed not or unlikely to be related to study medication.
The most common adverse events (overall frequency > 2%), by treatment group, in the safety population
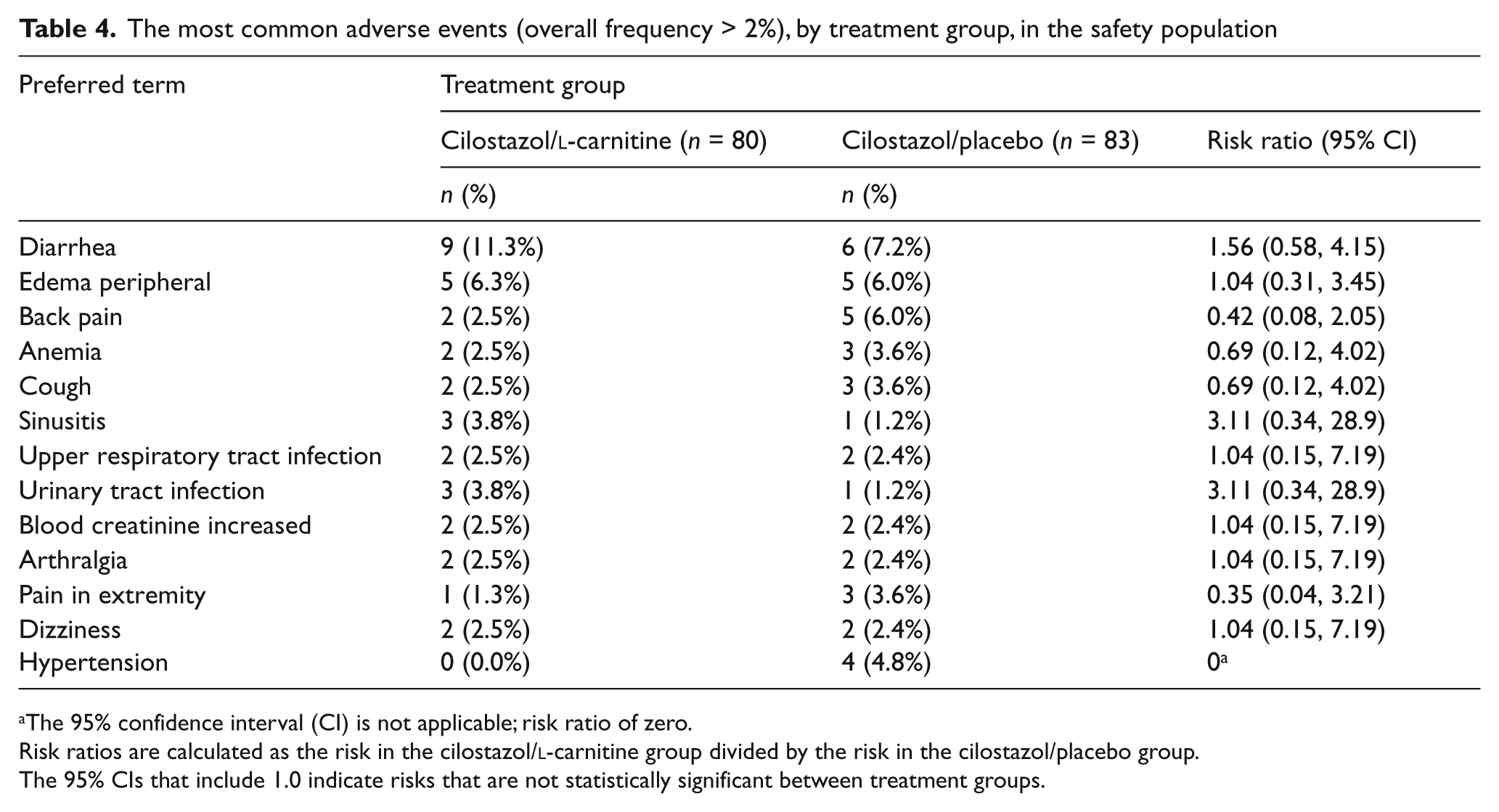
The 95% confidence interval (CI) is not applicable; risk ratio of zero.
Risk ratios are calculated as the risk in the cilostazol/l-carnitine group divided by the risk in the cilostazol/placebo group.
The 95% CIs that include 1.0 indicate risks that are not statistically significant between treatment groups.
Summary of all serious adverse events, by treatment group
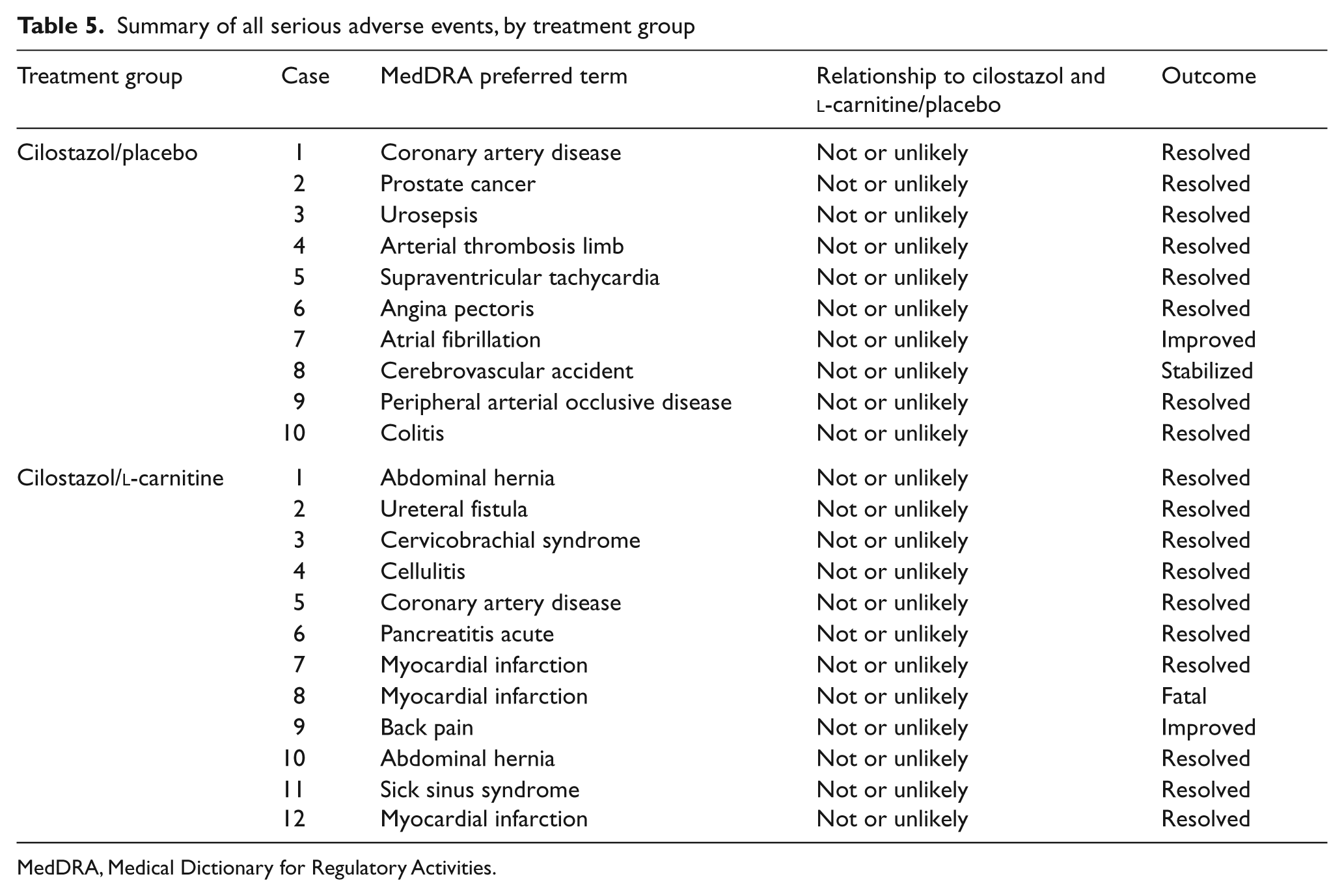
MedDRA, Medical Dictionary for Regulatory Activities.
Discussion
The present study of l-carnitine on a background of cilostazol versus cilostazol alone demonstrated a trend for efficacy for the combination of these agents in patients with IC due to PAD. We observed a greater numeric increase in PWT from baseline to day 180 among patients receiving cilostazol/l-carnitine than those receiving cilostazol/placebo in the mITT population, and a statistically significant increase in this endpoint in the per-protocol population. These findings were substantiated by trends or significant findings of efficacy in other relevant endpoints, including QOL measures. Furthermore, no safety concerns were manifest for cilostazol/l-carnitine relative to cilostazol/placebo.
Patients with PAD exhibit impaired oxygen uptake responses at the onset of exercise 29 despite unaltered blood flow at this early time point in exercise. 30 The acylcarnitine accumulation in plasma and skeletal muscle due to secondary carnitine deficiency in this setting has been inversely associated with exercise performance in PAD. 31 A reduced activity of key enzymes of the mitochondrial electron transport chain may also contribute to decreased capacity to generate ATP, and may be responsible (at least in part) for the reduced exercise performance of these patients. 32 On this basis, and in the context of the present findings, the addition of l-carnitine to a background therapy of cilostazol holds promise in the treatment of IC due to PAD.
No prior trials have been published studying combination therapies for IC in PAD, in which background therapy consisted of a standardized exercise regimen or cilostazol as established efficacious treatments. The magnitude of improvement in PWT detected at day 180 with the addition of l-carnitine to a conventional cilostazol regimen in the present trial is meaningful, representing an absolute increase of 17% (mean PWT change measured for cilostazol/l-carnitine 37.9% minus mean PWT change measured with cilostazol/placebo 20.9%). Furthermore, our results support prior observations of the potential efficacy of l-carnitine in clinical trials of PAD.11–15 Initial studies by Brevetti where l-carnitine was administered by oral 18 and intravenous 19 routes showed that supplementation with l-carnitine improved exercise performance in patients with IC. In the past decade, two pivotal multicenter, double-blinded, placebo-controlled RCTs have demonstrated the efficacy of extended carnitine therapy in PAD (delivered as propionyl-l-carnitine). A 12-month European RCT showed a significantly greater increase in maximum walking distance on a fixed treadmill, as well as a significantly improved QOL assessment, among patients treated with propionyl-l-carnitine 2 g daily when compared to those who received placebo. 13 The second trial, which employed a graded treadmill, demonstrated a significant increase in PWT following 6 months of therapy with carnitine 2 g daily versus placebo. 12 With regard to safety, in 2002 the FDA approved labeling changes for l-carnitine to include precautions for patients with severely compromised renal function or in end-stage renal disease patients on dialysis as administration may result in accumulation of the potentially toxic metabolites trimethylamine and trimethylamine-N-oxide, since these metabolites are normally excreted in the urine. No additional AEs have been clearly linked with carnitine supplementation.
Our findings for the cilostazol/placebo group (i.e. control subjects), while demonstrating a rather modest degree of improvement in PWT from baseline to day 180 (an arithmetic mean increase of 21%), is nevertheless consistent with a mean increase in PWT of 27% with dietary intervention, 33 and exceeds the observed mean increases in PWT of 15–17% for placebo in recent RCTs of IC in PAD.34,35 Moreover, the endpoint quality intervention program implemented in the present trial (which involves stringent site and equipment qualification, site endpoint training, and site endpoint evaluation visits for quality assurance), has been demonstrated to reduce the placebo response, 36 such that the effect of cilostazol and other interventions may be detected.
Limitations of the present trial are principally related to its sample size. Despite being among the largest RCTs of l-carnitine, its sample size resulted in insufficient power to detect a large intergroup difference in the primary endpoint. It is therefore possible that with a larger sample size, a statistically significant benefit with respect to the primary efficacy endpoint may have been detected in the mITT population. This limitation was largely mitigated by the control of variance in the primary endpoint of PWT, which (with a standard deviation of 3.75 minutes across all time points in the mITT population) is among the lowest yet reported in PAD trials. Furthermore, when the primary efficacy outcome is considered together with secondary efficacy findings (including a statistically significant benefit of the main efficacy endpoint in the per-protocol analysis), a strong signal of efficacy is suggested. A second potential limitation is the lack of a pure placebo arm (i.e. no background cilostazol). We believe that this is overcome by the fact that l-carnitine was studied in a placebo-controlled fashion in this trial, in which a background of cilostazol was used as the established mainstay of pharmacological therapy, given meta-analysis findings3,4 supporting its efficacy from numerous previous placebo-controlled RCTs. Lastly, given the predominance of Caucasian males in the study population, and although this is consistent with broader PAD incidence data, future trials should seek to target females and non-Caucasians more strategically.
In conclusion, treatment with cilostazol/l-carnitine for 6 months in PAD patients with IC appears safe and efficacious in improving PWT compared to cilostazol alone, with a beneficial effect that is magnified in the per-protocol population relative to the mITT population. These findings support the design of a definitively powered trial of oral l-carnitine 1 g twice-daily in combination with cilostazol 100 mg twice-daily as a potentially efficacious agent for IC in the PAD population, where treatment options are currently limited and no therapies addressing the primary metabolic defect in muscle metabolism have been approved by regulatory agencies.
Footnotes
Acknowledgements
The authors thank Pete Hanson of CPC for conducting the statistical analyses, and both Pete Hanson and Kelli Engelhardt for input during manuscript revision. CPC Clinical Research independently designed, executed, analyzed, and interpreted the findings of the trial.
ECLECTIC Trial Investigators consisted of the following: M Adams, Radiant Research–Salt Lake City; F Agnone, Internal Medicine Physicians Associates; D Benson, DMI Healthcare Group, Inc.; Y Chi, Ochsner Medical Center; A Comerota, Jobst Vascular Institute; B Cutler, University of Massachusetts Medical Center; D Dawson, University of California at Davis Vascular Center; L Egbujobi, Beloit Hospital; S Harlin, Pensacola Research Consultants, Inc.; P Hartsell, Clinical Trials of Texas, Inc.; W Herzog, Cardiovascular Specials of Central Maryland, PA; K Illig, University of Rochester Medical Center; B Jorgensen, Tatum Ridge Internal Medicine; G Lefebvre, Meridian Research; J Martinez, Peripheral Vascular Associates; B Molk, Aurora Denver Cardiology Associates–Aurora; NC Morcos, Apex Research Institute; K Morris, Durham VA Medical Center; M Moursi, Central Arkansas Veteran’s Healthcare System; B Nolan, Dartmouth-Hitchcock Medical Center; D Schumacher, Radiant Research–Columbus; D VanHamersveld, Sacramento Heart and Vascular Research Center; N Vijay, Aurora Denver Cardiology Associates–Denver; W Zhou, VA Palo Alto Health Care System.
Unrestricted grant support for the trial was provided to Dr Hiatt/CPC Clinical Research by Otsuka, Inc.
Support for other claudication trials has been provided to Dr Hiatt/CPC Clinical Research by the following pharmaceutical companies: Kowa, Diffusion, and Cytokinetics. The authors declare no other potential conflicts of interest.