
Abstract
Inflammation is critical for atherosclerosis development and may be a target for risk-reduction therapy. In experimental studies, activation of the inflammatory regulator, nuclear factor kappa B (NFlB), contributes to endothelial activation and reduced nitric oxide production. We treated patients with coronary artery disease with sulfasalazine, an inhibitor of NFκB, and placebo in a randomized, double-blind, crossover study design. Brachial artery flow-mediated dilation (FMD) and digital vascular function were measured at baseline and after each 6-week treatment period. Of the 53 patients enrolled in the crossover study, 32 (age 60 ± 10, 22% female) completed all the visits, with a high rate of study withdrawal due to gastrointestinal side effects. In a subset of 10 participants, we compared the effects of 4 days of sulfasalazine treatment (n = 5) to no treatment (n = 5) on NFκB-regulated gene expression in peripheral blood mononuclear cells. Tumor necrosis factor α-stimulated expression of CD69 and NFlB subunit p50 was significantly blunted after 4 days of sulfasalazine treatment but not after no treatment. However, FMD and digital vasodilator response did not significantly change from baseline with long-term sulfasalazine treatment. Short-term sulfasalazine inhibited NFlB activity; however, long-term treatment was poorly tolerated and did not improve endothelial function. Our findings suggest that sulfasalazine therapy is not the optimal anti-inflammatory treatment for reversing endothelial dysfunction in cardiovascular disease. Further studies are warranted to investigate the potential for NFlB inhibition to reduce cardiovascular risk.
Introduction
Abnormal endothelial function contributes to the pathogenesis of cardiovascular events in patients with clinical atherosclerosis.1–3 Systemic inflammation is a central mediator of all phases of atherogenesis including disruption of endothelial function.4,5 Novel anti-inflammatory therapies may have promise as a treatment strategy for cardiovascular risk reduction. 6
Prior evidence links inflammatory activation to altered endothelial phenotype leading to loss of nitric oxide bioactivity.5,7 Inflammatory cytokines induce endothelial expression of pro-inflammatory and prothrombotic factors that may contribute to cardiovascular risk. 8 There is considerable interest in the transcription factor, nuclear factor kappa B (NFκB), as a regulator of endothelial function.9,10 In animal models and in humans with cardiovascular risk factors, endothelial NFκB activation is associated with impaired vasodilator function.11–13
Sulfasalazine is a well-established anti-inflammatory drug that has been shown to inhibit the activation of NFκB. 14 A related compound, salsalate, has been shown to increase flow-mediated dilation (FMD) in obese humans; 13 however, the effects of NFκB inhibition in the setting of established clinical atherosclerotic disease remain unknown. Therefore, we conducted a randomized, double-blind, placebo-controlled, crossover study of sulfasalazine treatment to test the hypothesis that NFκB inhibition would improve vascular function in patients with stable coronary artery disease.
Methods
Study participants
We enrolled patients with stable coronary artery disease diagnosed by coronary angiography or a history of myocardial infarction. We excluded patients with unstable angina or myocardial infarction within 2 weeks of enrollment, allergy to sulfa-containing drugs, treatment with drugs that may interact with sulfasalazine (Coumadin, digoxin, phenytoin, methenamines, probenecid, sulfinpyrazones, oral hypoglycemic agents), immunosuppressive therapy, glucose-6-phosphate dehydrogenase deficiency, or active medical illness. The study protocol was approved by the Boston Medical Center Institutional Review Board and all participants provided written informed consent. The randomized portion of the study was registered on line (http://www.clinicaltrials.gov/, NCT00554203).
Sulfasalazine treatment and study design
We employed a randomized, double-blind, placebo-controlled, crossover study design. Treatment order was assigned using computer-generated randomization. As shown in Figure 1, after an initial screening visit, participants made four study visits. After the baseline study visit, participants were treated with sulfasalazine (Azulfidine EN; Pharmacia, New York, NY, USA) or placebo gelatin capsules for 6 weeks. During each active treatment period, participants were treated with one capsule of sulfasalazine or placebo twice daily for 1 week (1000 mg/day) then two capsules twice daily (2000 mg/day) for the remaining 5 weeks. The selected dose of sulfasalazine has been shown to have anti-inflammatory properties in rheumatoid arthritis. 15 After the initial treatment period, there was a 2-week rest period between treatments and then participants crossed over to the second treatment (sulfasalazine or placebo).
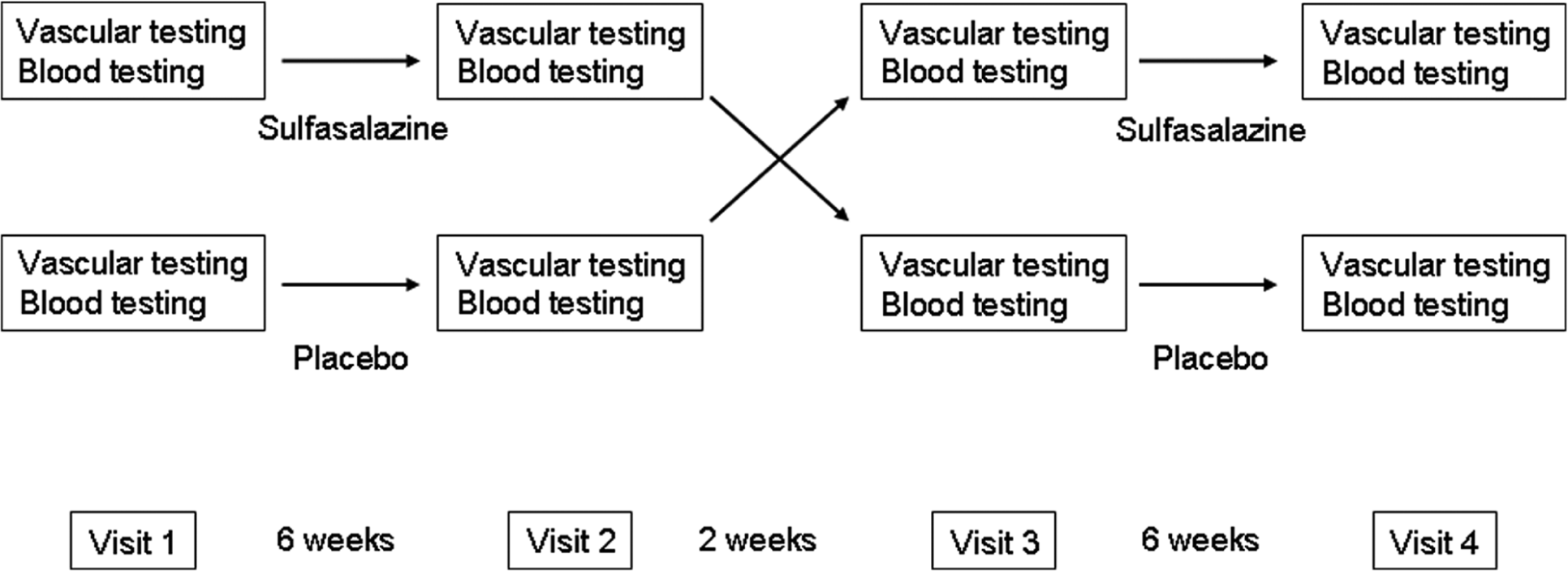
Study design. For the crossover study, participants were randomized to sulfasalazine treatment first (top row) or placebo first (bottom row) and received the assigned treatment for 6 weeks (one 500 mg or placebo pill twice daily for 1 week, then two 500 mg or placebo pills twice daily for 5 weeks). After a 2-week rest period, the participants crossed over to the alternate treatment.
Participants refrained from smoking and fasted after midnight on the morning of each study. Study participants were also instructed to stop vasoactive medications for the 24-hour period prior to each study visit. Participants took the study medications twice daily with the last dose on the morning of the study visit. We performed vascular testing and collected blood for evaluation of biomarkers at each study visit.
Vascular testing
We measured conduit vessel vasodilator function by assessing flow-mediated and nitroglycerin-mediated dilation of the brachial artery as previously described. 16 Briefly, high-resolution ultrasound was used to measure brachial artery diameter and Doppler flow at rest and following a 5-minute cuff occlusion of the upper arm to induce reactive hyperemia. To evaluate microvessel function in the finger vessels, we simultaneously performed digital pulse amplitude tonometry (EndoPAT; Itamar Medical Ltd, Caesarea, Israel). As we have previously described, we expressed the peripheral arterial tonometry (PAT) hyperemic response as the log-transformed ratio of the pulse amplitude in the test finger in the 90–120 second interval after cuff release to the resting pulse amplitude divided by the corresponding ratio in the contralateral control finger (PAT ratio). 17 We measured endothelium-independent brachial artery vasodilation following sublingual administration of nitroglycerin (0.4 mg). The nitroglycerin portion of the testing was omitted if the participant had a prior adverse reaction to nitrates, systolic blood pressure less than 100 mmHg, or use of phosphodiesterase type-5 medications.
Biochemical and inflammatory marker testing
Total cholesterol, high-density lipoprotein (HDL)-cholesterol, triglycerides and glucose were measured using enzymatic methods in the Boston Medical Center Clinical Laboratory. Low-density lipoprotein (LDL)-cholesterol was calculated using the Friedewald formula. 18 Insulin was measured using the immunochemiluminometric methodology. Serum C-reactive protein was determined with a high-sensitivity nephelometric method. 19 Serum interleukin-6, p-selectin, monocyte chemotactic protein-1, and soluble intracellular adhesion molecule-1 were measured using commercially available kits (R&D Systems, Inc., Minneapolis, MN, USA).
Assessment of NFlB activity in peripheral blood mononuclear cells
To examine the effect of sulfasalazine treatment on NFκB activation in peripheral leukocytes, we enrolled an additional 10 participants with coronary artery disease. We treated five participants for 4 days with sulfasalazine 1000 mg twice daily and five participants received no treatment. Blood was collected before and after the 4 days in a Ficoll separation tube (CPT™; Becton Dickinson, Franklin Lakes, NJ, USA) and centrifuged at 1650 g for 30 minutes. Isolated mononuclear cells were washed in Hank’s balanced salt solution (HBSS) and treated with 0, 1, or 10 ng/ml tumor necrosis factor alpha (TNFα) (R&D Systems, Inc.) in HBSS for 2 hours in a rotating incubator at 37°C. Total RNA was isolated using the RNeasy Mini Kit (Qiagen, Inc., Alameda, CA, USA) and RNA was reverse transcribed to complimentary DNA using TaqMan® Reverse Transcription Reagents (Applied Biosystems, Carlsbad, CA, USA). Quantitative real-time PCR (TaqMan Gene Expression Assays; Applied Biosystems) was performed to measure the expression of genes regulated by NFκB, including NFκB subunit p50 (NFκB1, Hs00765730_m1) and CD69 (Hs00934033_m1). Results were interpreted by the relative quantity method (ΔΔCt) using glyceraldehyde-3-phosphate dehydrogenase (GAPDH) as a loading control.
Statistical analysis
Analyses were completed using SPSS for Windows, version 12.1 (SPSS Inc.) and a two-sided p-value < 0.05 was considered statistically significant. Values are reported as mean ± SD, unless otherwise indicated. We compared the baseline clinical characteristics of the two treatment order groups (sulfasalazine first and placebo first) using the unpaired t-test or chi-square test as appropriate. We evaluated the effect of treatment order on each endpoint by two-way repeated-measures analysis of variance (ANOVA). We evaluated the effect of treatment by paired t-test comparing the after sulfasalazine to the after placebo conditions. We evaluated the gene expression response to TNFα before and after sulfasalazine treatment using two-way repeated-measures ANOVA.
Based on prior data from our laboratory, the sample size of 32 participants who completed all four study visits in a crossover design provides greater than 90% power (alpha = 0.05) to detect a 2.0 percentage point change (e.g. 7–9%) in brachial artery FMD, the primary endpoint of the study.
Results
Study participants
As shown in Figure 2, 53 participants qualified for the study and were randomized. There was a high withdrawal rate (n = 21, 40%), which was predominantly related to gastrointestinal symptoms during active treatment (n = 11). The clinical characteristics of the participants are shown in Table 1. The participants in the two treatment order groups (sulfasalazine first and placebo first) were similar, with a high prevalence of cardiovascular risk factors and a high rate of concurrent cardiovascular medications. There were no differences in baseline clinical characteristics between the participants who completed the study and the participants who withdrew.
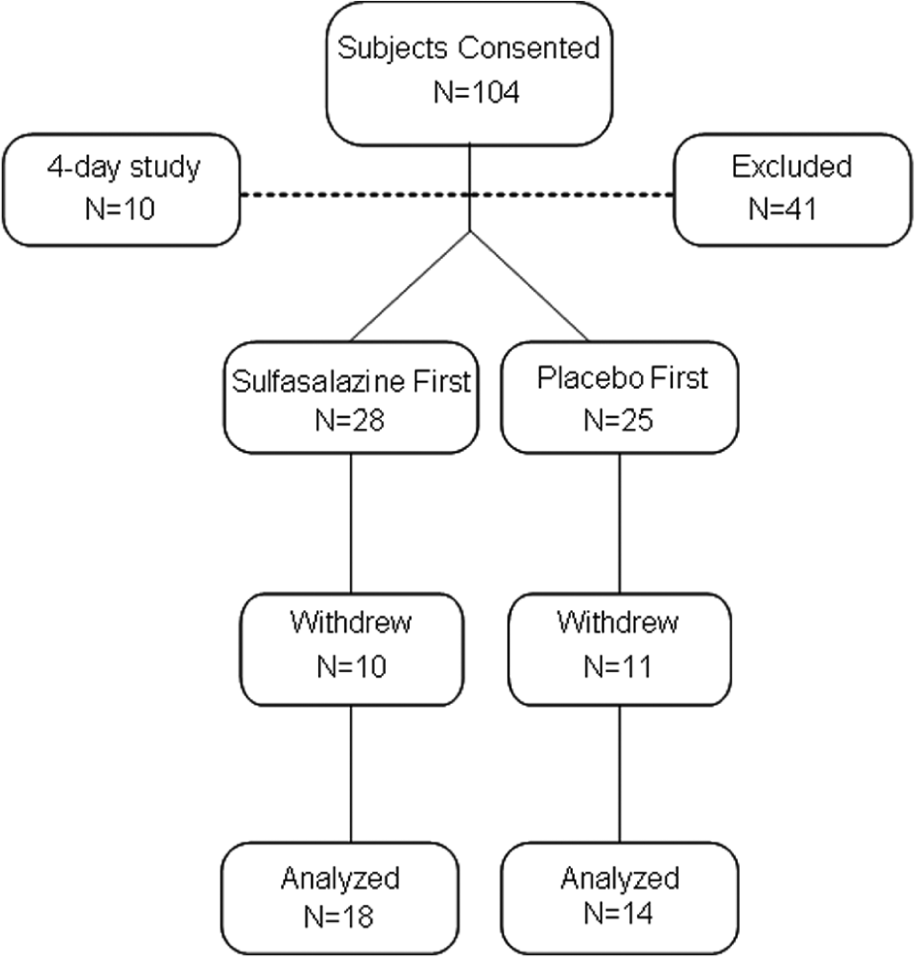
Consolidated Standards of Reporting Trials (CONSORT) flow diagram.
Baseline clinical characteristics – randomized crossover study
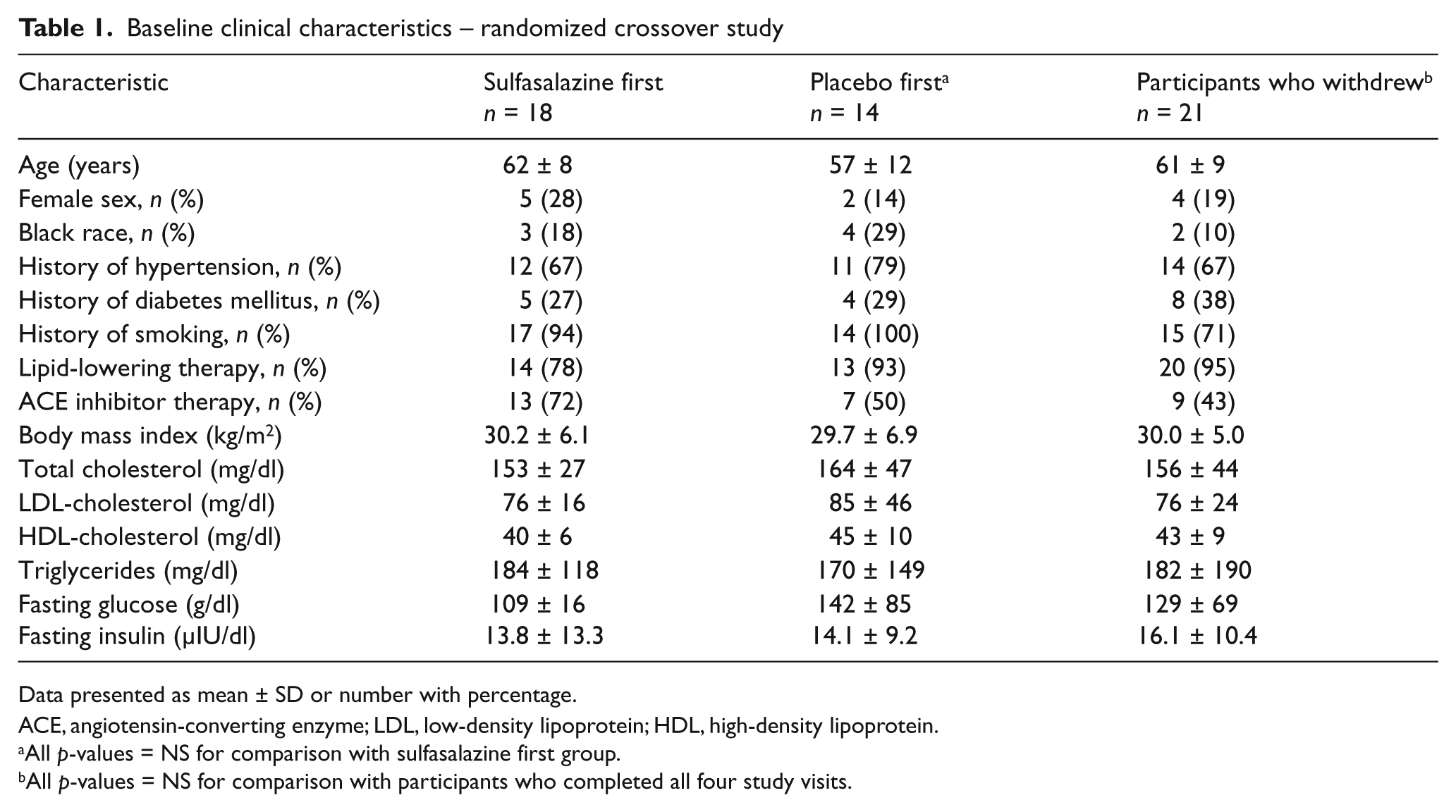
Data presented as mean ± SD or number with percentage.
ACE, angiotensin-converting enzyme; LDL, low-density lipoprotein; HDL, high-density lipoprotein.
All p-values = NS for comparison with sulfasalazine first group.
All p-values = NS for comparison with participants who completed all four study visits.
Effect of sulfasalazine on vascular function
We evaluated whether sulfasalazine treatment has an effect on vascular function in coronary artery disease patients. As shown in Table 2, we found no effect of sulfasalazine on brachial artery FMD, a measure of conduit vessel endothelial function, or on PAT hyperemic response, a measure of small vessel vasodilator function. In addition, we found no effect of sulfasalazine on blood pressure, hyperemic response, or arterial diameter. There was no evidence of an effect of treatment order for any of the vascular measures.
Effects of sulfasalazine on vascular function
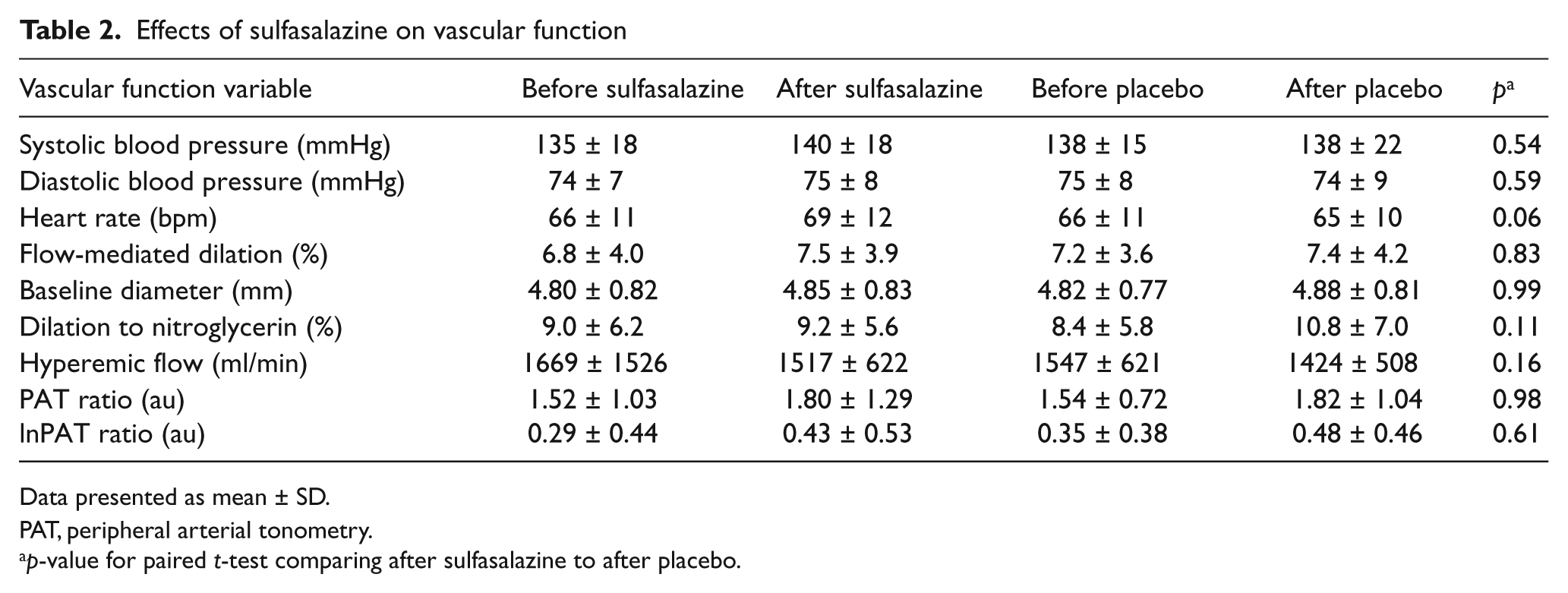
Data presented as mean ± SD.
PAT, peripheral arterial tonometry.
p-value for paired t-test comparing after sulfasalazine to after placebo.
In secondary analyses, we evaluated the effect of sulfasalazine on FMD in selected subgroups of participants. We found no effect of sulfasalazine in subgroups including: participants with HOMA-IR below the group median; participants without diabetes; participants with C-reactive protein less than 10 mg/l; participants with FMD below the median at the baseline visit; men, women, or white race (data not shown). In the group of seven black participants, FMD was lower with sulfasalazine treatment compared to placebo (5.4 ± 4.9% compared to 8.7 ± 5.8%, p = 0.015). The apparent difference in this subgroup likely reflects the small number of individuals included.
Effect of sulfasalazine on biomarkers
As shown in Table 3, we observed no effect of sulfasalazine treatment on serum lipids, glucose, or insulin levels. There were no effects of sulfasalazine on multiple systemic markers of inflammation.
Effects of sulfasalazine on biochemical profile
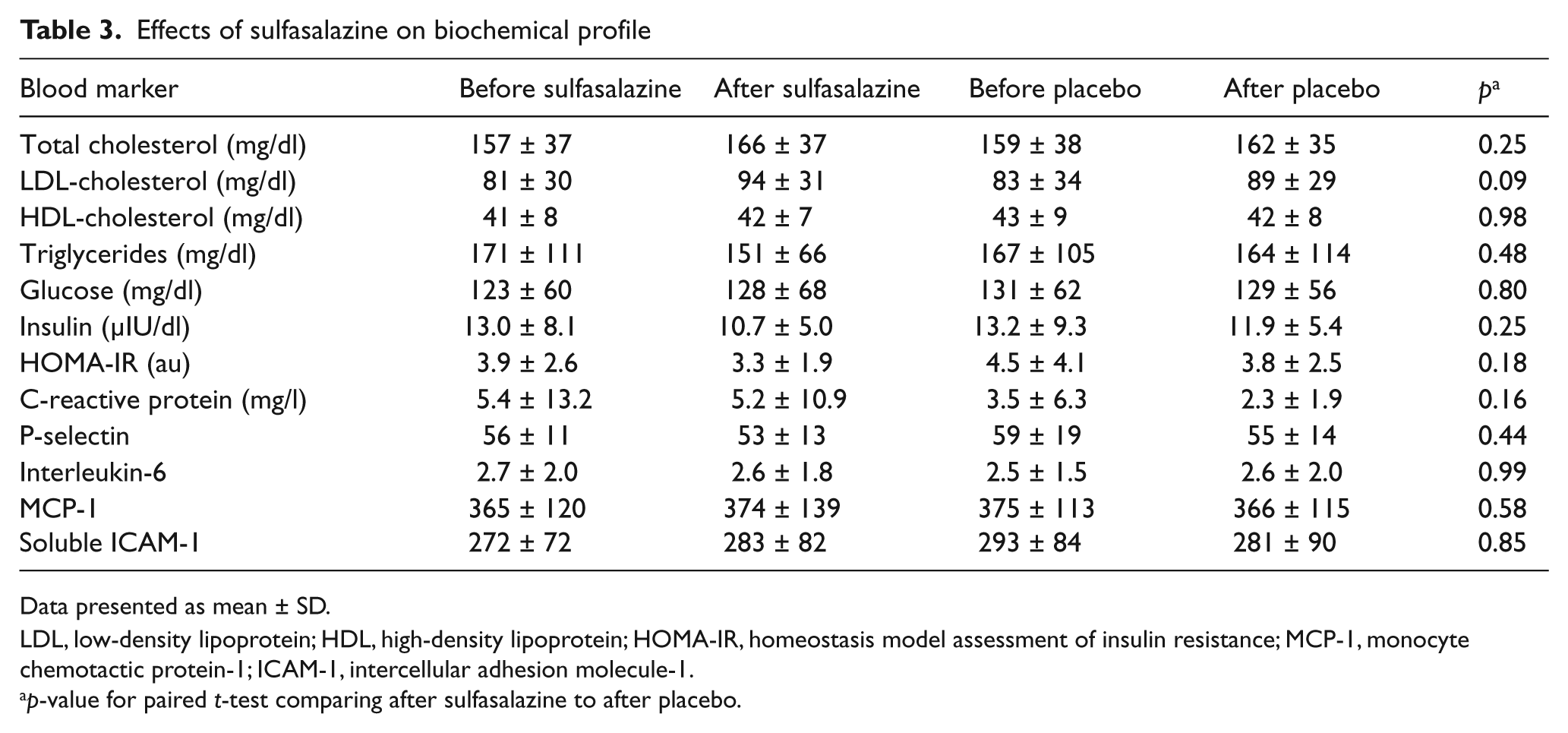
Data presented as mean ± SD.
LDL, low-density lipoprotein; HDL, high-density lipoprotein; HOMA-IR, homeostasis model assessment of insulin resistance; MCP-1, monocyte chemotactic protein-1; ICAM-1, intercellular adhesion molecule-1.
p-value for paired t-test comparing after sulfasalazine to after placebo.
Effect of sulfasalazine on NFκB activation
To gain support of an effect of sulfasalazine on NFκB activity in patients with coronary artery disease (n = 10), we collected peripheral leukocytes before and after 4 days with (n = 5) or without (n = 5) sulfasalazine treatment. As shown in Figure 3, we observed that sulfasalazine treatment markedly blunted the induction of NFκB-regulated gene expression in response to TNFα, consistent with inhibition of NFκB activation. Participants who received no sulfasalazine treatment had a similar response to TNFα stimulation at baseline compared to after 4 days for both NFκB1 (p = 0.13) and CD69 (p = 0.14). There was a significant difference in the effect of sulfasalazine as compared to no intervention on TNFα-stimulated NFκB1 and CD69 gene expression (p = 0.026 and p = 0.009, comparing sulfasalazine to no treatment groups by repeated-measures ANOVA).
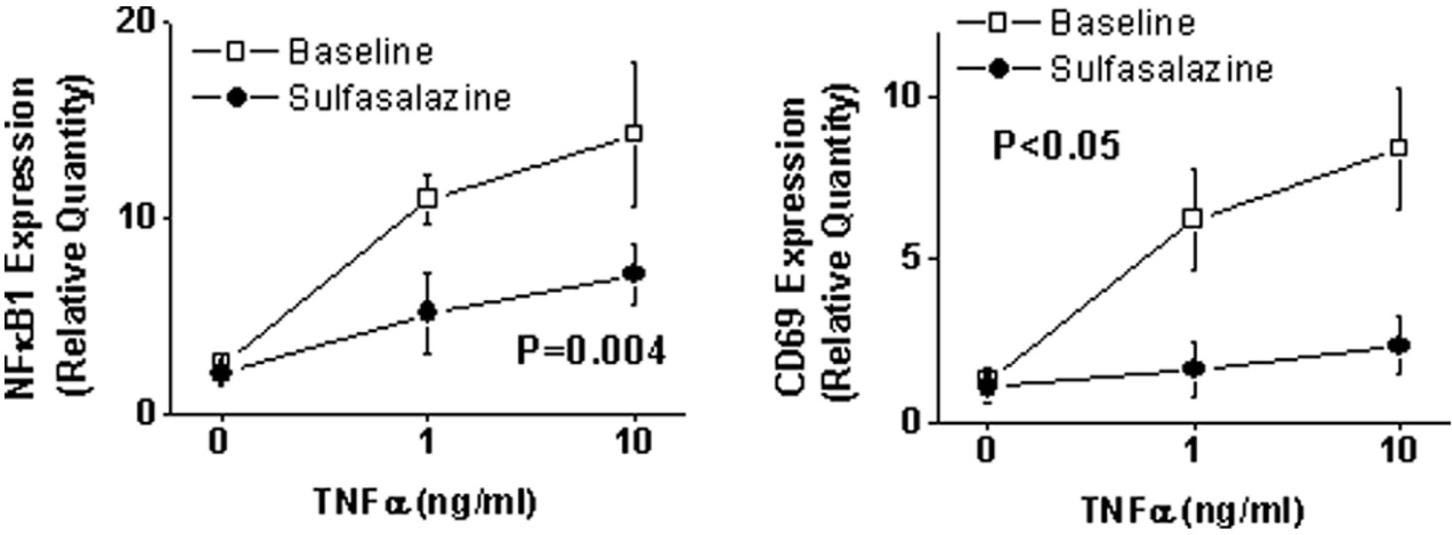
Effect of sulfasalazine on NFκB-mediated inflammatory activity. Peripheral blood mononuclear cells were isolated as described in the Methods section before and after 4 days of sulfasalazine treatment (1000 mg twice daily) in five patients with coronary artery disease. Expression levels of NFκB-regulated genes were assessed with quantitative PCR after 2 hours of treatment with TNFa at 0, 1, and 10 ng/ml. The p-value shown is for comparison of overall gene expression before and after sulfasalazine treatment.
Discussion
The present crossover study investigated whether NFκB inhibition with sulfasalazine has a favorable effect on vascular function in stable coronary artery disease. Short-term treatment with sulfasalazine reduced NFκB activity as evidenced by a blunted induction of NFκB-regulated genes following TNFα stimulation of freshly isolated peripheral blood mononuclear cells. In contrast, we observed no effect of long-term sulfasalazine treatment on systemic inflammatory markers. Inhibition of NFκB with sulfasalazine was not associated with an improvement of endothelial function as measured in both conduit and microvessels. Sulfasalazine was poorly tolerated in our group of patients with established atherosclerosis as the study withdrawal rate was greater than 40%. Taken together, our findings do not support a vascular benefit of sulfasalazine in patients with chronic coronary artery disease.
Activation of NFκB contributes to inflammation in coronary artery disease. NFκB is a transcription factor that controls the expression of proinflammatory genes relevant to atherogenesis including cytokines, chemokines and cell adhesion molecules.20,21 Human atherosclerotic plaques contain activated NFκB, and in animal models NFκB activity localizes to atherosclerosis-prone regions.22,23 Patients with unstable coronary syndromes have evidence of NFκB activation in circulating blood monocytes that is associated with a higher risk of future cardiovascular events.24,25 Thus, inhibition of NFκB-mediated inflammatory responses has been proposed as a strategy to ameliorate risk in coronary artery disease.25,26
Prior studies establish non-acetylated salicylic acids, including sulfasalazine, as inhibitors of NFκB activation. Sulfasalazine acts by preventing phosphorylation and subsequent degradation of the NFκB inhibitory subunit, IκB.14,27–29 In cultured cells, salicylates inhibit endothelial activation and reduce intracellular adhesion molecule expression. 30 We provide evidence that short-term oral sulfasalazine administration blunts NFκB activity in patients with coronary artery disease. In freshly isolated peripheral blood mononuclear cells, there was a marked reduction in TNFα-induced expression of inflammatory genes regulated by NFκB following sulfasalazine treatment. Interestingly, the anti-inflammatory effect of sulfasalazine observed in peripheral blood mononuclear cells did not correspond with a change in circulating markers of inflammation. In this regard, our findings are consistent with two previous human studies in HIV patients and obese adults that showed no effect of salsalate treatment on systemic inflammatory markers.13,31 Thus, salicylates appear to reduce intracellular inflammatory responses without altering circulating inflammatory biomarker levels.
Several lines of evidence implicate inflammation in the pathogenesis of endothelial dysfunction. 5 In experimental models, inflammatory mediators limit nitric oxide bioavailability, in part through reduced endothelial nitric oxide synthase expression.32–34 In human studies, systemic inflammatory markers associate with impaired endothelial vasodilator function.5,35 Acute inflammatory states induce the development of transient endothelial dysfunction.36,37 There is evidence that NFκB activation may have particular relevance to impaired vascular function observed in the presence of cardiovascular risk factors. Endothelial cells isolated from older and obese individuals demonstrate higher NFκB activation that is associated with reduced FMD.11,13
The current study showed no favorable effects of sulfasalazine treatment on endothelial function. In coronary artery disease patients, both conduit artery and digital vessel FMD were unchanged by sulfasalazine treatment. Our findings are discordant with prior work indicating beneficial effects of salicylates on endothelial function in select samples. In a study of older obese adults without diabetes or clinical cardiovascular disease, 4 days of salsalate therapy improved brachial artery FMD and reduced endothelial cell NFκB activation. 13 In an uncontrolled pilot study, salsalate treatment for 8 weeks improved FMD in patients with HIV disease. 31
Multiple potential explanations exist for the apparent discrepancy. Clinical characteristics of our study participants may have reduced the efficacy of anti-inflammatory treatment. Our study sample included patients in a more advanced phase of disease with established clinical cardiovascular disease. It is possible that endothelial dysfunction in chronic disease may be more resistant to improvement with NFκB inhibition. Alternatively, our participants were intensively treated with secondary prevention interventions as evidenced by well-controlled lipid levels. Sulfasalazine therapy may have limited effect in medically optimized patients. In addition, it is possible that salsalate has greater efficacy for improving endothelial function than sulfasalazine. The limited effect of sulfasalazine may relate to poor tolerability due to frequent gastrointestinal side effects.
Study limitations
The current study has a number of limitations. The high rate of participant withdrawal reduced our overall sample size for analysis. However, the available sample size provided adequate power to detect a change in FMD similar in magnitude to that observed in prior intervention studies. It is possible that concurrent medication use may have modulated the effect of sulfasalazine; though the study design with a crossover methodology limits the influence of confounding. We did not measure serum levels of sulfasalazine or its metabolites, limiting our ability to confirm adherence. The selected sulfasalazine dose was based on studies in rheumatoid arthritis and we did demonstrate an effect on circulating cells; however, the effective dose required to influence the endothelium may be higher. Given the poor tolerability at the dose used in the present study, using a higher dose would have limited clinical utility. It is possible that the dissociation of the NFκB activation results and the vascular function and serum markers relate to the different duration of treatment. Sulfasalazine is a pharmacologic inhibitor of NFκB that may have other non-specific anti-inflammatory effects. We used an upper arm cuff placement to induce hyperemia; the results may differ with a forearm cuff placement. Finally, the short-term study evaluating NFκB activation in leukocytes was not randomized. However, all cell analyses were performed in a manner blinded to the study condition.
Conclusions
In summary, treatment with sulfasalazine reduced NFκB activation but did not improve endothelial function in patients with coronary artery disease. Sulfasalazine was poorly tolerated in our sample of older individuals taking multiple concurrent medications. Our findings counter the hypothesis that sulfasalazine might have the potential to reverse endothelial dysfunction in established atherosclerotic disease. The optimal strategy for mitigating the adverse effects of chronic inflammation in patients with atherosclerosis remains an ongoing area of investigation. Further studies are needed to evaluate the clinical efficacy of NFκB inhibition as a strategy to lower cardiovascular risk.
Footnotes
Acknowledgements
We wish to thank Dr Peter Merkel for serving as the safety monitor for this study.
This work was supported by NIH grants HL074097, HL084213, and HL083269. Drs Tabit, Vita, and Hamburg are supported by the Boston University Medical Center Leadership Program in Vascular Medicine (K12 HL083781). Dr Vita is supported by grants from the National Institutes of Health (HL083801, HL081587, and HL75795). Dr Hamburg is supported by a grant from the National Institutes of Health (HL102299). Dr Dohadwala was supported by T32 HL007224.
The authors have no conflicts of interest.