
Abstract
Background:
The clinical relevance of serum glial fibrillary acidic protein (sGFAP) concentration as a biomarker of MS disability progression independent of acute inflammation has yet to be quantified.
Objective:
To test whether baseline values and longitudinal changes in sGFAP concentration are associated with disability progression without detectable relapse of magnetic resonance imaging (MRI) inflammatory activity in participants with secondary-progressive multiple sclerosis (SPMS).
Methods:
We retrospectively analyzed longitudinal sGFAP concentration and clinical outcome data from the Phase 3 ASCEND trial of participants with SPMS, with no detectable relapse or MRI signs of inflammatory activity at baseline nor during the study (n = 264). Serum neurofilament (sNfL), sGFAP, T2 lesion volume, Expanded Disability Status Scale (EDSS), Timed 25-Foot Walk (T25FW), 9-Hole Peg Test (9HPT), and composite confirmed disability progression (CDP) were measured. Linear and logistic regressions and generalized estimating equations were used in the prognostic and dynamic analyses.
Results:
We found a significant cross-sectional association between baseline sGFAP and sNfL concentrations and T2 lesion volume. No or weak correlations between sGFAP concentration and changes in EDSS, T25FW, and 9HPT, or CDP were observed.
Conclusion:
Without inflammatory activity, changes in sGFAP concentration in participants with SPMS were neither associated with current nor predictive of future disability progression.
Keywords
Introduction
Glial fibrillary acidic protein (GFAP), first discovered in 1971, 1 is a type III intermediate filament found in astrocytes. 2 Driven in part by the potential role of astrocytes in MS pathology 3 and the correlations found between serum glial fibrillary acidic protein (sGFAP) and neurofilament light chain (NfL),4,5 there has been increasing interest in exploring the utility of sGFAP concentration as a biomarker in neurological diseases, 6 including in MS. 7 This interest has been accelerated through the development of novel technologies such as single-molecule arrays (Simoa), which permit the measurement of sGFAP concentration in peripheral blood. 8 Areas of focus have included its potential correlation with clinical and radiological measures of inflammatory activity, clinical disability scores,4,9–11 and alterations in normal-appearing white matter. 12 A consistent finding has been that sGFAP concentrations are higher in progressive forms of the disease and may be correlated with disease progression longitudinally,4,5,9,13–17 although this has very recently been disputed. 18 Nevertheless, these findings are suggestive of a potential value of sGFAP that is distinct from NfL measurement, as they imply that sGFAP concentration may capture disease biology in MS that may be independent of or less dependent on acute neuroinflammation, the latter being more characteristic of the earlier stages of the disease. In this context, we recently demonstrated in a large clinical trial data set of participants with secondary-progressive multiple sclerosis (SPMS) that serum neurofilament (sNfL) was not a dynamic biomarker of disability progression that occurred independently of acute inflammation, 19 and findings for sNfL as a biomarker of progression in MS remain inconclusive. 20
Given the absence of a robust peripheral biomarker of disability progression in MS, 7 and to mitigate the potential confounding impact of acute inflammation on sGFAP concentrations, we set out in this study using data from ASCEND, 21 a large clinical trial of participants with SPMS, to determine the potential value of sGFAP measurements as biomarkers of disability progression in the absence of relapses and magnetic resonance imaging (MRI) evidence of inflammatory activity.
Methods
Study design
ASCEND (NCT01416181) was a multicenter, randomized, double-blind, placebo-controlled, Phase 3 study exploring the effect of natalizumab (TYSABRI®) on disease progression in SPMS. Details of the study design, outcomes, ethical considerations, and patient consent requirements have been previously described. 21
sGFAP concentration was measured in samples collected at baseline and at Weeks 24, 48, 72, and 96 by the Simoa technology on an HD-1 Analyzer using the sGFAP Discovery Kit, as described by the manufacturer (Quanterix, Billerica, MA, USA). 8 Standardized T2-weighted and pre- and post-contrast T1-weighted MRIs were collected every 24 weeks starting from baseline to end of study, 19 and MRI lesions were assessed by a centralized MRI reading center (NeuroRx, Montreal, Canada). Slowly expanding lesions (SELs) were identified as areas of non-enhancing T2-lesions pre-existing at baseline that showed constant and concentric local expansion from baseline to Week 108, as measured on T1-weighted and T2-weighted MRI as previously described. 22 In this study, the absence of acute inflammatory activity was defined as no baseline or post-baseline (at Weeks 24, 48, 72, 96, and 108) gadolinium-enhancing lesions on T1-weighted post-contrast MRI, no post-baseline new or enlarging lesions T2 lesions during the same time points, and no clinical relapses during the study period.
Expanded Disability Status Scale (EDSS), Timed 25-Foot Walk (T25FW), and 9-Hole Peg Test (9HPT) on either hand assessments were performed at baseline and every 12 weeks through Week 108. Composite confirmed disability progression (CDP) was defined as meeting one or more of the following three criteria: an increase of ⩾1.0 points from a baseline EDSS score ⩽5.5 or an increase of ⩾0.5 point from a baseline score ⩾6.0, an increase of ⩾20% from baseline in T25FW time, or an increase of ⩾20% from baseline in 9HPT time (on either hand). 9 Progression was confirmed at ⩾6 months. Individual CDP was defined similarly as above but for each criterion (EDSS, T25FW, and 9HPT either hand) separately. Confirmed disability improvement was defined similarly as above but with decreases instead of increases.
Statistical analyses
Repeated measure analysis of variance (ANOVA) was used separately for each treatment group to compare if sGFAP concentrations changed significantly across time. Importantly, ANOVA was used to test equality of means across all time points; therefore, a rejection of the null hypothesis (denoted by a significant p-value) signifies that at least one time point’s mean is different from the rest, without specifying which time point. Spearman correlation coefficients were used to evaluate the univariate correlation between sNfL and sGFAP concentrations measured concurrently. To cope with the skewness of the sGFAP distribution, statistical analyses were performed using log2-transformed sGFAP data unless specified otherwise.
Analyses of baseline sGFAP
The predictive modeling of baseline sGFAP as an outcome variable using baseline characteristics (age, sex, MS duration, T2 lesion volume, log2-transformed sNfL) as independent variables was evaluated by a linear regression model. The associations between baseline sGFAP as an independent variable versus T2 and T1 lesion volumes as outcome variables (i.e. baseline and percentage change from baseline to Week 96 in T2 and T1 lesion volumes and in volume of SELs vs non-SELs lesion subtypes) were analyzed by linear regression models. Logistic regressions were used to study the association between baseline sGFAP versus CDP and confirmed disability improvement. Analyses were performed for each treatment group separately and to the pooled cohort as a sensitivity analysis. False discovery rate was used to adjust p-values for multiple testing. Unless specified otherwise, all analyses controlled for six baseline variables: age, sex, MS duration, T2 lesion volume, sNfL, and EDSS score.
Dynamic analyses of sGFAP
Changes in sGFAP concentration were defined as percentage changes in log2-transformed sGFAP relative to log2-transformed sGFAP concentration at baseline. Correlations between changes in sGFAP and changes in disability assessment scores post-baseline were investigated separately for each variable (EDSS, T25FW, 9HPT) and treatment group (natalizumab and placebo). For each disability assessment score, one generalized estimating equations model was applied to four correlated outcomes: percentage changes in sGFAP concentration and percentage changes in the disability assessment score, both measured at two time periods—baseline to Week 48, and Week 48 to Week 96, with each subject as a cluster while adjusting for the six aforementioned baseline covariates, the corresponding baseline assessment score, and the time period indicator (baseline to Week 48, Week 48 to Week 96). The assumed correlation structure of the generalized estimating equations was unstructured, and robust variance estimators were used. The association between composite score progression and changes in sGFAP was analyzed using logistic regression separately by treatment group and time intervals (baseline to Week 48, Week 48 to Week 96) adjusting for the same six baseline covariates. Analyses were performed in R 4.2.2, and significance level was 0.05.
Results
We acquired sGFAP concentrations of 264 ASCEND intent-to-treat participants with no inflammatory activity who had sGFAP concentrations available from each of the five time points (Weeks 0, 24, 48, 72, 96); 177 (67%) had been assigned to the natalizumab arm and 87 (33%) to the placebo arm. The flow of number of participants is summarized in Supplemental Table 1, and baseline characteristics are summarized in Table 1. At baseline, the mean (standard deviation (SD)) age was 49.2 (6.4) years, and there were 84 (32%) males. Median (25th–75th percentile) EDSS score was 6.0 (5.0–6.5) at baseline. Figure 1 shows the distribution of sGFAP concentrations across time by treatment group. Overall, sGFAP concentrations stayed relatively constant across time in this cohort (p = 0.848 for the natalizumab group and p = 0.609 for the placebo group, indicating time points are not significantly different). The placebo group had relatively higher median concentrations than the natalizumab group, but this was not statistically significant at any time point (all p > 0.05). sGFAP and sNfL concentrations were weakly correlated at Weeks 0, 48, and 96 (estimated Spearman’s correlations: 0.21, 0.30, and 0.17 for natalizumab group at each time point and 0.31, 0.33, and 0.27 for placebo group, respectively; Supplemental Figure 1).
Baseline characteristics of the ASCEND non-inflammatory cohort.
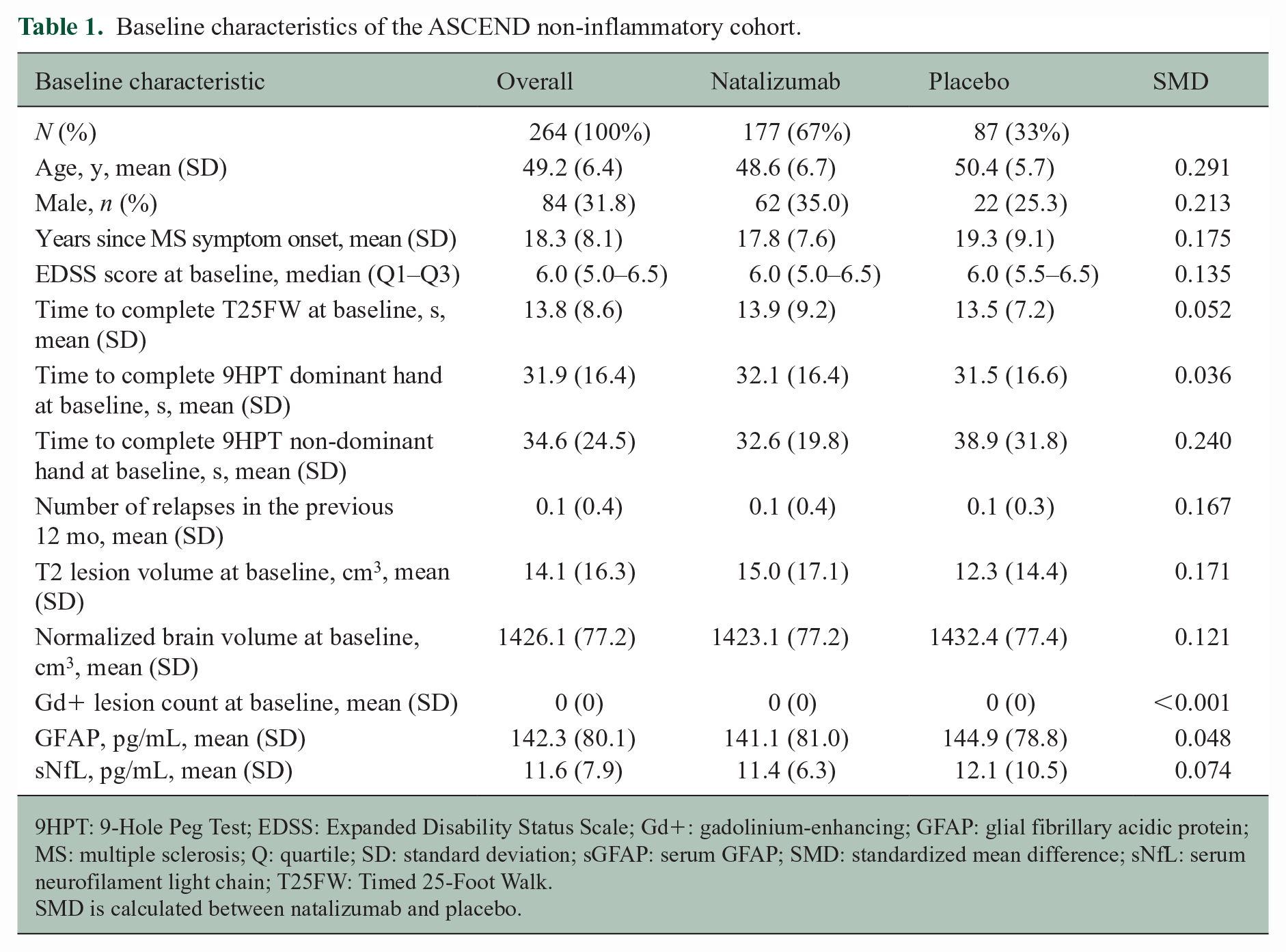
9HPT: 9-Hole Peg Test; EDSS: Expanded Disability Status Scale; Gd+: gadolinium-enhancing; GFAP: glial fibrillary acidic protein; MS: multiple sclerosis; Q: quartile; SD: standard deviation; sGFAP: serum GFAP; SMD: standardized mean difference; sNfL: serum neurofilament light chain; T25FW: Timed 25-Foot Walk.
SMD is calculated between natalizumab and placebo.
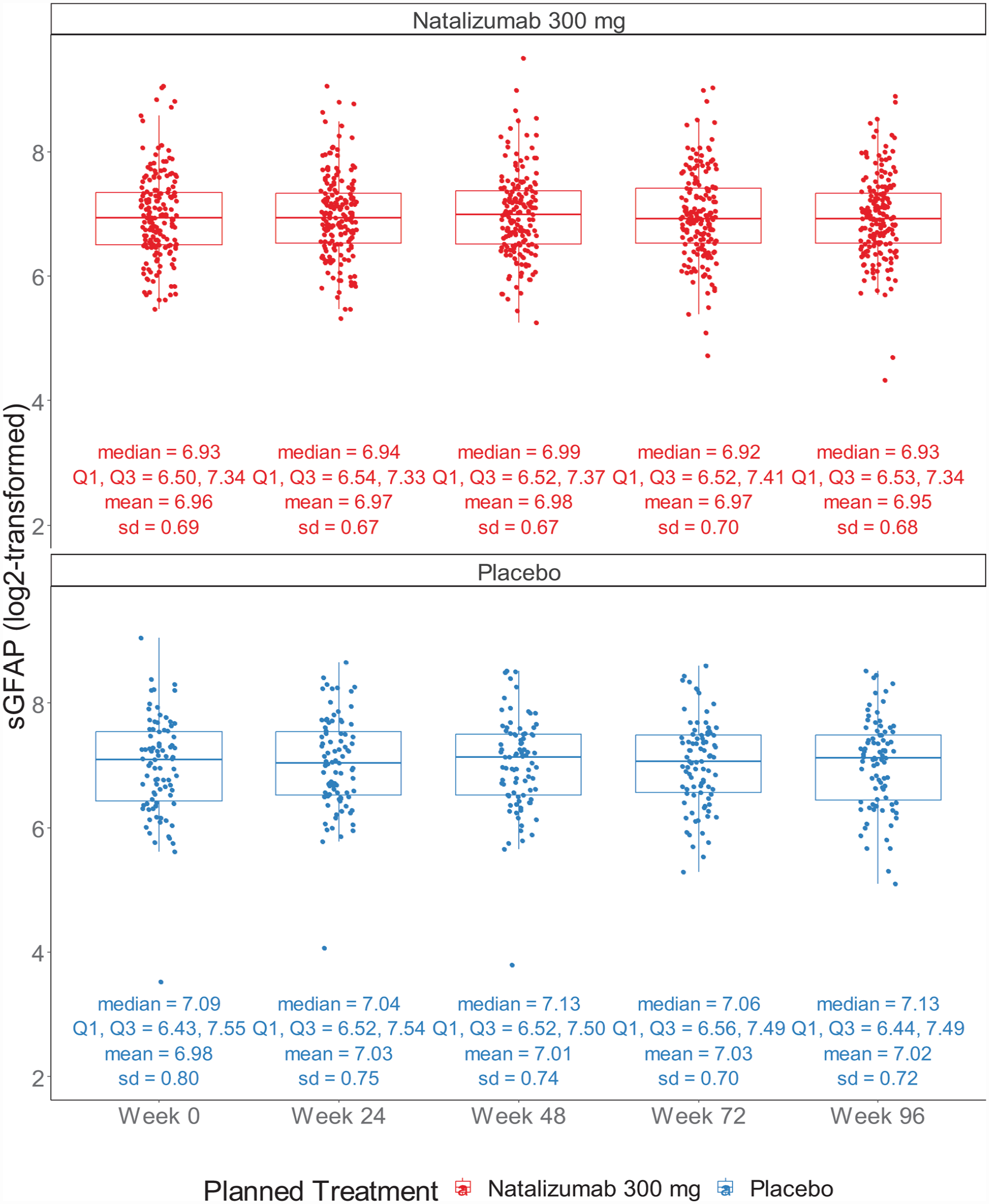
Distribution of sGFAP concentrations across time by treatment group.a,b
Analyses of baseline sGFAP
Baseline sGFAP was found to be significantly associated with baseline T2 lesion volume and sNfL in the pooled cohort (p < 0.001 for both), in the natalizumab group (p < 0.001 and p = 0.014, respectively), and baseline sNfL in the placebo group (p = 0.002) (Supplemental Table 2). No significant associations were found between baseline sGFAP concentration and T2 or T1 (SELs or non-SELs) lesion volume at baseline or between baseline sGFAP and T2 or T1 (SELs or non-SELs) lesion volume changes across the 96 weeks of study in natalizumab, placebo, or pooled cohorts (p > 0.05 for all) (Supplemental Table 3). Between baseline and Week 96, composite CDP occurred in 51% of the natalizumab population and 46% of the placebo cohort (Figure 2). No significant association was found between baseline sGFAP and composite or individual CDP and confirmed improvement, except for EDSS confirmed improvement in the natalizumab group where the odds ratio (95% confidence interval (CI)) of improvement for every 10% increase in baseline sGFAP concentration was 1.07 (1.00, 1.15) (Table 2). This endpoint was no longer significant in the sensitivity analysis of the pooled cohort (Supplemental Table 4), and the results were relatively similar in the sensitivity analysis of not adjusting for baseline sNfL (Supplemental Table 5).
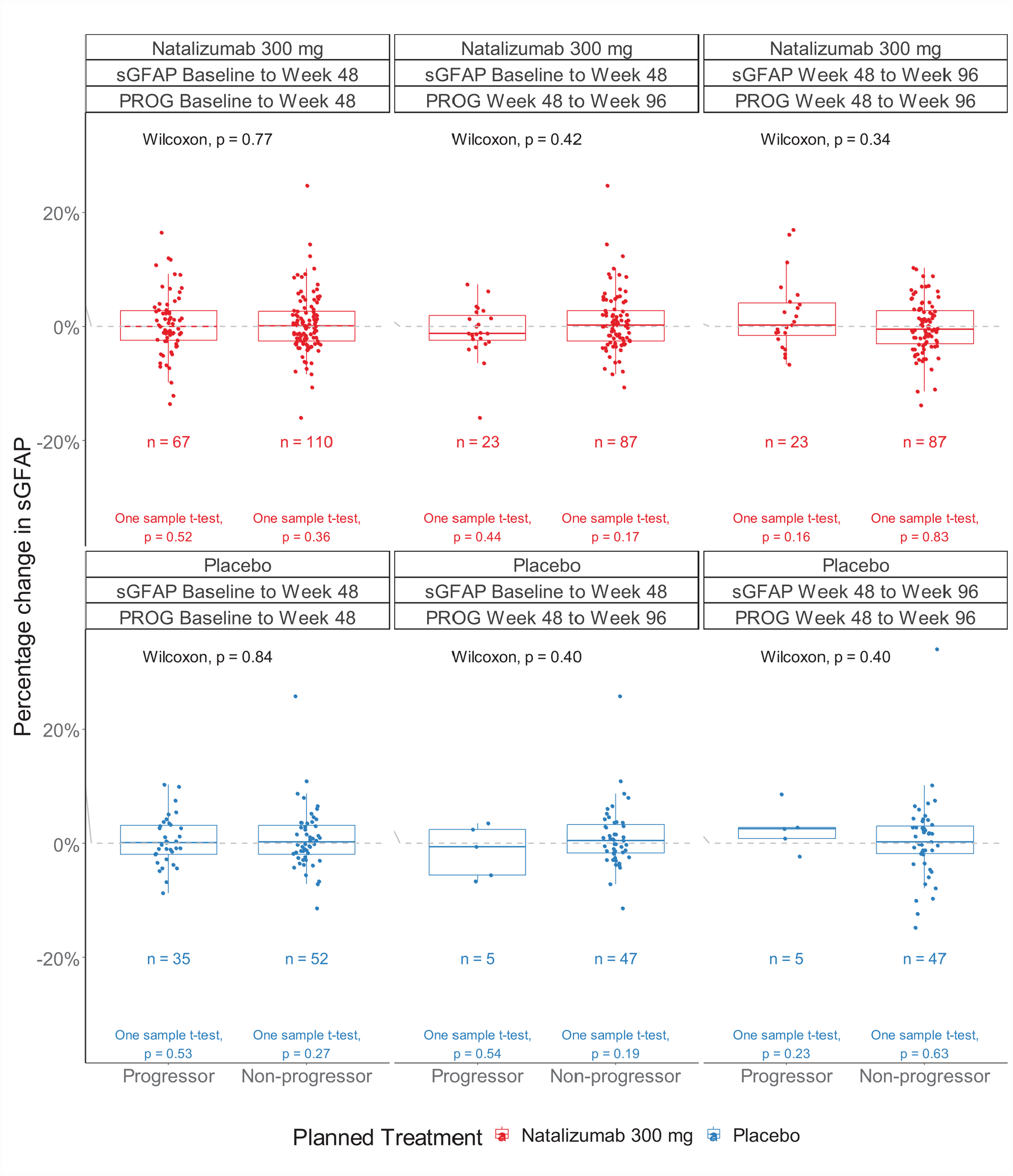
Distribution of changes in sGFAP concentrations by progressor status, time epochs (baseline to Week 48, Week 48 to Week 96), and treatment group.a,b,c,d
Estimated odds ratio (95% CI) of composite CDP after BL up to Week 96 and improvement for every 10% increase in BL sGFAP concentration. a
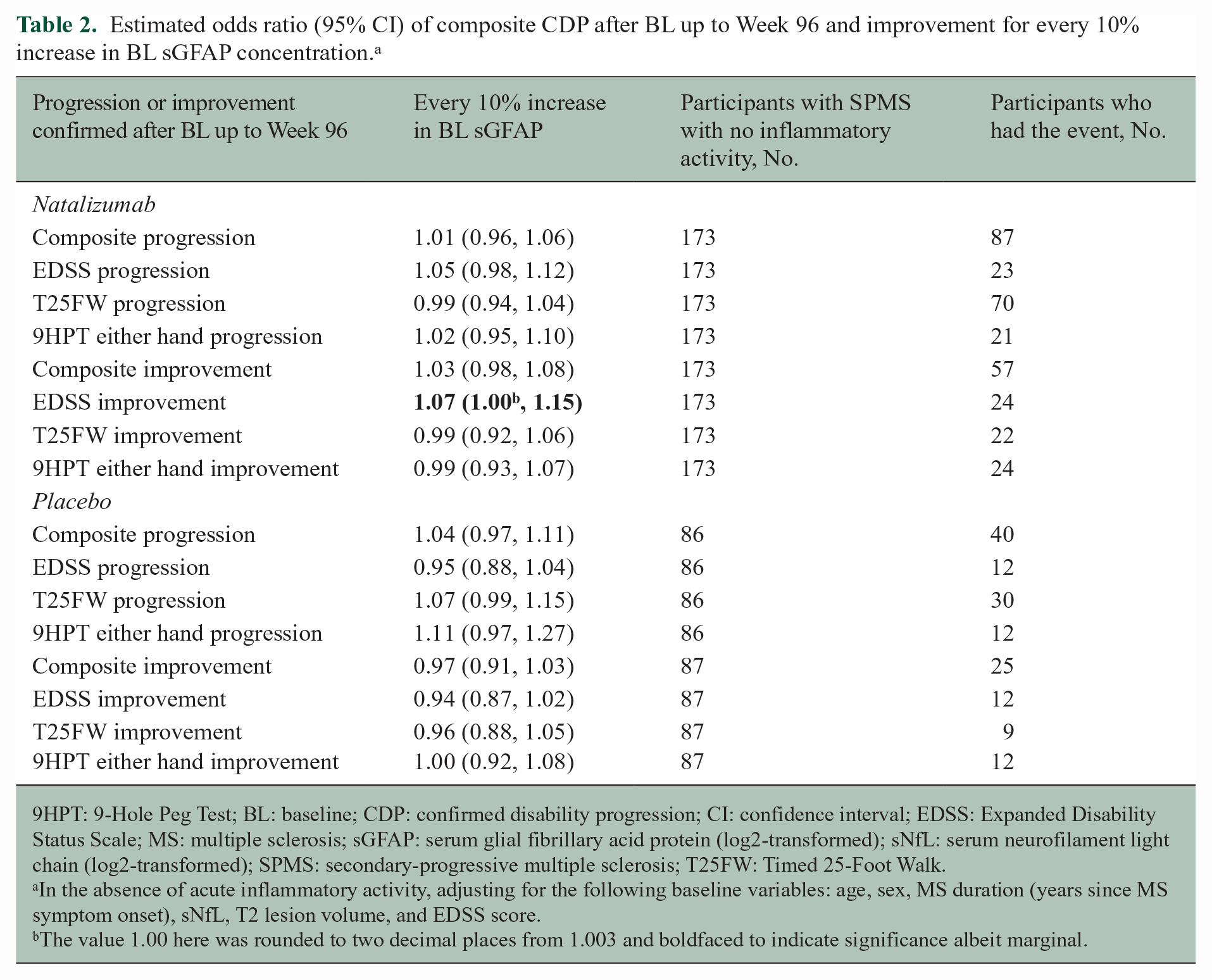
9HPT: 9-Hole Peg Test; BL: baseline; CDP: confirmed disability progression; CI: confidence interval; EDSS: Expanded Disability Status Scale; MS: multiple sclerosis; sGFAP: serum glial fibrillary acid protein (log2-transformed); sNfL: serum neurofilament light chain (log2-transformed); SPMS: secondary-progressive multiple sclerosis; T25FW: Timed 25-Foot Walk.
In the absence of acute inflammatory activity, adjusting for the following baseline variables: age, sex, MS duration (years since MS symptom onset), sNfL, T2 lesion volume, and EDSS score.
The value 1.00 here was rounded to two decimal places from 1.003 and boldfaced to indicate significance albeit marginal.
Dynamic analyses of sGFAP
There was no correlation or only weak correlation between changes in sGFAP concentration over the treatment period of 2 years and current or future changes in EDSS, T25FW, and 9HPT in both treatment groups (Table 3). All correlation coefficients were within 0.05 in absolute values with 95% CIs including 0, indicating no evidence of significant correlation. When stratifying individuals by their CDP status, there was no evidence to suggest that a change in sGFAP concentration was a significant independent predictor of current progression or of future disability progression over the subsequent 48 weeks (Figure 2 and Table 4).
Estimated correlation (95% CI) between change in sGFAP concentration and change in disability assessment score. a .
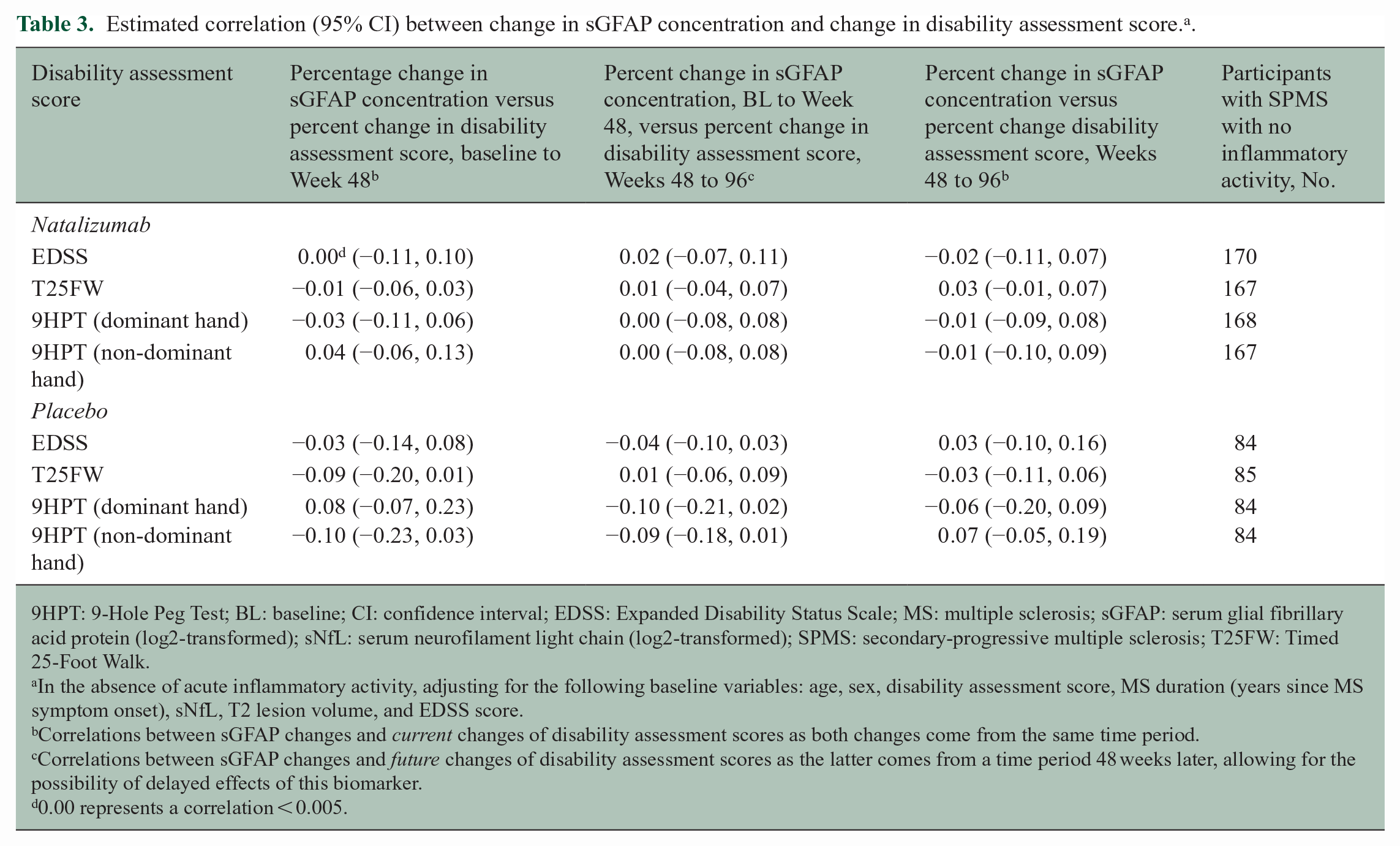
9HPT: 9-Hole Peg Test; BL: baseline; CI: confidence interval; EDSS: Expanded Disability Status Scale; MS: multiple sclerosis; sGFAP: serum glial fibrillary acid protein (log2-transformed); sNfL: serum neurofilament light chain (log2-transformed); SPMS: secondary-progressive multiple sclerosis; T25FW: Timed 25-Foot Walk.
In the absence of acute inflammatory activity, adjusting for the following baseline variables: age, sex, disability assessment score, MS duration (years since MS symptom onset), sNfL, T2 lesion volume, and EDSS score.
Correlations between sGFAP changes and current changes of disability assessment scores as both changes come from the same time period.
Correlations between sGFAP changes and future changes of disability assessment scores as the latter comes from a time period 48 weeks later, allowing for the possibility of delayed effects of this biomarker.
0.00 represents a correlation < 0.005.
Estimated odds ratio (95% CI) of composite CDP for every 10% increase in sGFAP concentration. a
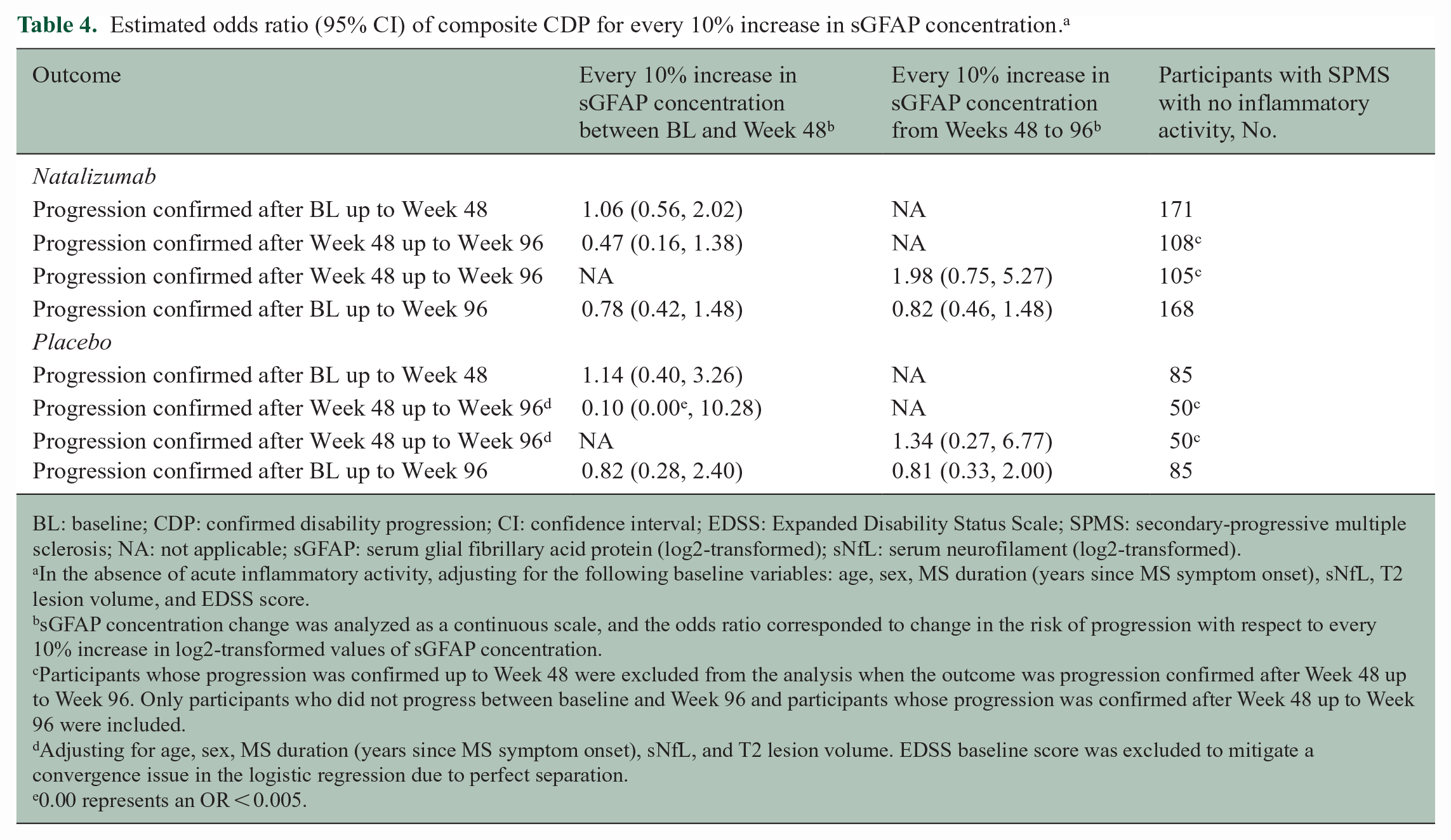
BL: baseline; CDP: confirmed disability progression; CI: confidence interval; EDSS: Expanded Disability Status Scale; SPMS: secondary-progressive multiple sclerosis; NA: not applicable; sGFAP: serum glial fibrillary acid protein (log2-transformed); sNfL: serum neurofilament (log2-transformed).
In the absence of acute inflammatory activity, adjusting for the following baseline variables: age, sex, MS duration (years since MS symptom onset), sNfL, T2 lesion volume, and EDSS score.
sGFAP concentration change was analyzed as a continuous scale, and the odds ratio corresponded to change in the risk of progression with respect to every 10% increase in log2-transformed values of sGFAP concentration.
Participants whose progression was confirmed up to Week 48 were excluded from the analysis when the outcome was progression confirmed after Week 48 up to Week 96. Only participants who did not progress between baseline and Week 96 and participants whose progression was confirmed after Week 48 up to Week 96 were included.
Adjusting for age, sex, MS duration (years since MS symptom onset), sNfL, and T2 lesion volume. EDSS baseline score was excluded to mitigate a convergence issue in the logistic regression due to perfect separation.
0.00 represents an OR < 0.005.
Discussion
This retrospective study of a large cohort of participants with SPMS explored the dynamics of sGFAP concentration in the absence of acute inflammation and its association with contemporaneous and future disability progression in participants with SPMS over 2 years of a clinical trial. Similar to previous publications,9,10 we found a significant cross-sectional association between baseline sGFAP concentration and sNfL concentration and T2 lesion volume. However, sGFAP concentrations were not predictive of future disability or T2 lesion volume change and did not change dynamically with disability progression.
There are several reasons why measuring sGFAP concentration might not have been able to capture the dynamics of MS disability progression in this cohort. First, unlike sNfL, 23 sGFAP concentrations did not change significantly during the study period and were therefore less likely to be sensitive to changes in progression. Nevertheless, in contrast to other studies,4,5,9,13–16 we were also unable to demonstrate that baseline sGFAP concentrations were prognostic of future disability progression. The latter finding may be due to the short duration setting of clinical trials, the absence of acute inflammation in our cohort (which plays some part in clinical disability progression), or the lack of sensitivity and indeed low concentrations of the biomarker when detected in the periphery (as opposed to cerebrospinal fluid). Interestingly, in contrast to our findings, a recent study in a real-world cohort was able to demonstrate that GFAP may be prognostic of disability progression independent of inflammation; this cohort had a longer follow-up time period, and the participants had a lower EDSS on average. 24 Second, disease progression in MS that occurs in the absence of acute inflammation is likely to be mediated (either individually or in combination) by a chronic neuron-axonal degeneration due to chronic demyelination and the failure of remyelination, chronic active demyelination related to smoldering inflammation, or even by environmental factors (e.g. lack of exercise, smoking, or depression). In this context, an astrocytic biomarker alone may not be able to capture the complex combination of insidious drivers of a slow and progressive pathophysiological process, in particular if astrocytic proliferation only plays a limited part. Third, the expression and function of sGFAP is worth further consideration. Despite the moderate correlations with sNfL concentrations, sGFAP is solely expressed in astrocytes. First isolated from old MS plaques, expression is predominantly observed in fibrous gliosis (i.e. in the final glial scar). 1 Interestingly, in our cohort, we confirmed an association between total T2 lesion volume and sGFAP concentration. Although some pathologically defined chronic lesions in MS are active or smoldering, this may represent a minority.25,26 Indeed, radiologically defined chronic active lesion correlates, such as SELs, only represent a very small proportion of the total T2 lesion volume. 22 In our cohort, this proportion was 4.6%. Whereas the relative density of sGFAP concentration in chronic active versus inactive lesions has not been characterized, one could speculate that much of the burden of peripheral sGFAP seen in participants with MS may not be coming from lesion subtypes that drive progression in MS but rather from inactive plaque tissue that cannot be reliably distinguished from other types of T2 lesions on MRI. As such, concentrations of sGFAP may simply represent overall acquired lesion burden from disease onset. Worth noting also is that the role of astrocytes in driving progression in MS still remains controversial. 27
This study has limitations. It focused on clinical disability outcomes and was restricted to clinical trial participants with SPMS and may not be generalizable to other MS populations. The absence of acute inflammation in the placebo cohort may have represented a more benign cohort whereas in the treated arm, this would have been a combination of treatment effect and benign disease. Radiological measures of progression, such as brain atrophy, were not investigated, and our smaller sample size in the placebo population could have underpowered the analysis in this group due to the impact of outliers on the results. The definition of a “non-inflammatory” population might not be rigorously accurate due to the difficulty in discerning acute inflammation that occurs in pre-existing areas of inflammation or in chronic active lesions. The short trial duration did not allow us to model the predictive value of sGFAP concentration on longer-term disability progression.
sGFAP concentration, as measured by Simoa, did not appear to be a prognostic or dynamic biomarker of disability progression over 2 years in MS. Further work is required to understand better the relative contribution of astrocytes in the pathology driving progression in MS and whether the source of sGFAP detected in the periphery is predominantly derived from active or inactive lesions.
Supplemental Material
sj-docx-1-msj-10.1177_13524585231176732 – Supplemental material for Glial fibrillary acidic protein and multiple sclerosis progression independent of acute inflammation
Supplemental material, sj-docx-1-msj-10.1177_13524585231176732 for Glial fibrillary acidic protein and multiple sclerosis progression independent of acute inflammation by Xiaotong Jiang, Changyu Shen, Charlotte E Teunissen, Mark Wessels, Henrik Zetterberg, Gavin Giovannoni, Carol M Singh, Bastien Caba, Colm Elliott, Elizabeth Fisher, Carl de Moor, Shibeshih Belachew and Arie R Gafson in Multiple Sclerosis Journal
Supplemental Material
sj-docx-2-msj-10.1177_13524585231176732 – Supplemental material for Glial fibrillary acidic protein and multiple sclerosis progression independent of acute inflammation
Supplemental material, sj-docx-2-msj-10.1177_13524585231176732 for Glial fibrillary acidic protein and multiple sclerosis progression independent of acute inflammation by Xiaotong Jiang, Changyu Shen, Charlotte E Teunissen, Mark Wessels, Henrik Zetterberg, Gavin Giovannoni, Carol M Singh, Bastien Caba, Colm Elliott, Elizabeth Fisher, Carl de Moor, Shibeshih Belachew and Arie R Gafson in Multiple Sclerosis Journal
Supplemental Material
sj-docx-3-msj-10.1177_13524585231176732 – Supplemental material for Glial fibrillary acidic protein and multiple sclerosis progression independent of acute inflammation
Supplemental material, sj-docx-3-msj-10.1177_13524585231176732 for Glial fibrillary acidic protein and multiple sclerosis progression independent of acute inflammation by Xiaotong Jiang, Changyu Shen, Charlotte E Teunissen, Mark Wessels, Henrik Zetterberg, Gavin Giovannoni, Carol M Singh, Bastien Caba, Colm Elliott, Elizabeth Fisher, Carl de Moor, Shibeshih Belachew and Arie R Gafson in Multiple Sclerosis Journal
Supplemental Material
sj-docx-4-msj-10.1177_13524585231176732 – Supplemental material for Glial fibrillary acidic protein and multiple sclerosis progression independent of acute inflammation
Supplemental material, sj-docx-4-msj-10.1177_13524585231176732 for Glial fibrillary acidic protein and multiple sclerosis progression independent of acute inflammation by Xiaotong Jiang, Changyu Shen, Charlotte E Teunissen, Mark Wessels, Henrik Zetterberg, Gavin Giovannoni, Carol M Singh, Bastien Caba, Colm Elliott, Elizabeth Fisher, Carl de Moor, Shibeshih Belachew and Arie R Gafson in Multiple Sclerosis Journal
Supplemental Material
sj-docx-5-msj-10.1177_13524585231176732 – Supplemental material for Glial fibrillary acidic protein and multiple sclerosis progression independent of acute inflammation
Supplemental material, sj-docx-5-msj-10.1177_13524585231176732 for Glial fibrillary acidic protein and multiple sclerosis progression independent of acute inflammation by Xiaotong Jiang, Changyu Shen, Charlotte E Teunissen, Mark Wessels, Henrik Zetterberg, Gavin Giovannoni, Carol M Singh, Bastien Caba, Colm Elliott, Elizabeth Fisher, Carl de Moor, Shibeshih Belachew and Arie R Gafson in Multiple Sclerosis Journal
Supplemental Material
sj-docx-6-msj-10.1177_13524585231176732 – Supplemental material for Glial fibrillary acidic protein and multiple sclerosis progression independent of acute inflammation
Supplemental material, sj-docx-6-msj-10.1177_13524585231176732 for Glial fibrillary acidic protein and multiple sclerosis progression independent of acute inflammation by Xiaotong Jiang, Changyu Shen, Charlotte E Teunissen, Mark Wessels, Henrik Zetterberg, Gavin Giovannoni, Carol M Singh, Bastien Caba, Colm Elliott, Elizabeth Fisher, Carl de Moor, Shibeshih Belachew and Arie R Gafson in Multiple Sclerosis Journal
Footnotes
Acknowledgements
The authors acknowledge that editorial support for the finalization of this manuscript was provided by Excel Scientific Solutions (Fairfield, CT, USA).
Author Contributions
X.J., A.R.G., C.S., and S.B. contributed to the concept and design. X.J., A.R.G., C.S., and S.B. contributed to the acquisition or interpretation of data. X.J., A.R.G., and C.S. contributed to the statistical analysis. X.J. and A.R.G. contributed to the drafting of the manuscript. All authors contributed to the critical revision of the manuscript for important intellectual content.
Declaration of Conflicting Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: A.R.G., X.J., C.S., C.M.S., B.C., E.F., C.d.M., and S.B. are employees of and hold stock/stock options in Biogen. C.T. has a collaboration contract with ADx Neurosciences and Quanterix, and performed contract research for AC-Immune, Axon Neurosciences, Biogen, Brainstorm Therapeutics, Celgene, EIP Pharms, Eisai, PeopleBio, Roche, Toyama, and Vivoryon. M.W. reports no disclosures. H.Z. has served at scientific advisory boards and/or as a consultant for AbbVie, Acumen, Alector, ALZPath, Annexon, Apellis, Artery Therapeutics, AZTherapies, CogRx, Denali, Eisai, NervGen, Novo Nordisk, Passage Bio, Pinteon Therapeutics, Red Abbey Labs, reMYND, Roche, Samumed, Siemens Healthineers, Triplet Therapeutics, and Wave, has given lectures in symposia sponsored by Cellectricon, Fujirebio, AlzeCure, Biogen, and Roche, and is a co-founder of Brain Biomarker Solutions in Gothenburg AB (BBS), which is a part of the GU Ventures Incubator Program, outside the submitted work. G.G. has received compensation for serving as a consultant or speaker for or has received research support from AbbVie, Aslan, Atara Bio, Biogen, BMS-Celgene, GlaxoSmithKline, GW Pharma, Janssen/Actelion, Japanese Tobacco, Jazz Pharmaceuticals, LifNano, Merck & Co., Merck KGaA/EMD Serono, Moderna, Novartis, Sanofi-Genzyme, Roche/Genentech, and Teva. C.E. has received speaker honoraria from EMD Serono and is an employee of NeuroRx Research.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was funded by Biogen (Cambridge, MA, USA). The authors had full editorial control of the manuscript and provided their final approval of all content.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.