
Abstract
While the major phenotypes of multiple sclerosis (MS) and relapsing–remitting, primary and secondary progressive MS have been well characterized, a subgroup of patients with an active, aggressive disease course and rapid disability accumulation remains difficult to define and there is no consensus about their management and treatment. The current lack of an accepted definition and treatment guidelines for aggressive MS triggered a 2018 focused workshop of the European Committee for Treatment and Research in Multiple Sclerosis (ECTRIMS) on aggressive MS. The aim of the workshop was to discuss approaches on how to describe and define the disease phenotype and its treatments. Unfortunately, it was not possible to come to consensus on a definition because of unavailable data correlating severe disease with imaging and molecular biomarkers. However, the workshop highlighted the need for future research needed to define this disease subtype while also focusing on its treatment and management. Here, we review previous attempts to define aggressive MS and present characteristics that might, with additional research, eventually help characterize it. A companion paper summarizes data regarding treatment and management.
Introduction
Multiple sclerosis (MS) is a serious, lifelong disabling disease with unpredictable outcomes. The clinical presentation and phenotype of MS are variable between patients and over time. It can encompass various degrees of severity. A minority of patients exhibit either a benign course with little disability accrual over time or an ‘aggressive’ course with frequent, severe relapses, incomplete recovery and rapidly accumulating and permanent disability. 1
The efficacy of available disease-modifying therapies (DMTs) for relapsing multiple sclerosis (rMS) and progressive (primary progressive MS (PPMS) and secondary progressive MS (SPMS)) have been insufficiently studied in aggressive disease courses. 2 For some available DMTs, regulatory approval and insurance coverage mandate that patients must be considered not responsive (based on ongoing clinical or radiological disease activity) or intolerant to first-line therapies before receiving access to more effective treatments. This strategy is debatable in patients with a more aggressive form of disease, in which the successful therapeutic window of opportunity may be narrower than for those with less aggressive disease. 3 Patients with aggressive MS may be better treated if given rapid access to therapies considered to be more effective at slowing or preventing disability accrual. This approach would decrease the probabilities of breakthrough disease with first-line DMTs. Therefore, early identification of subjects with aggressive disease at onset or with disease that becomes more aggressive over time is essential to both aid treatment recommendations and decisions in the clinic. In addition, having a unified definition of aggressive MS could be used as a tool to enrich enrolment and stratify subjects for prospective clinical trials for such disease phenotype. 4 In the current report, attempts to define aggressive/active MS are reviewed. The group was unable to come to consensus about a new, more data-driven definition. However, we provide a detailed description of clinical and paraclinical factors that are relevant to substantiate a definition, such as imaging, neuropathological and immunological findings, as well as biomarker and genetic factors that should be studied to determine possible associations with the clinical phenotype of aggressive MS.
Materials and methods
To explore aggressive MS, its definition and treatment, the European Committee for Treatment and Research in Multiple Sclerosis (ECTRIMS) held a focused workshop on aggressive MS in March 2018 in Brussels, Belgium, including 50 European, North and South American, Asian and Australian neurologists and other professionals with interest in the understudied subgroup of MS patients with aggressive disease. Presentations on the clinical, paraclinical and biological characteristics of aggressive MS were followed by discussions about current treatment strategies and recommendations for future research that will allow improved definitions of aggressive MS and more effective and tailored treatment of affected patients. To support information presented at the workshop and data which appeared after, while this manuscript was being prepared, we conducted literature searches in the English language using PubMed, applying the following search terms: ‘multiple sclerosis’ OR ‘MS’ AND ‘aggressive’ and ‘multiple sclerosis’ OR ‘MS’ AND ‘highly active’.
Results
Past and recent efforts to define aggressive/highly active/malignant MS
Previous efforts to characterize severe or aggressive MS focused on the group of patients with highly active disease (frequent and severe relapses, rapid worsening) and high inflammatory and neurodegenerative activity, generally those with poor prognosis and outcomes over relatively short periods of time (Table 1). These have been variously categorized as those with ‘malignant MS’, 5 ‘aggressive MS’ 10 or ‘highly active MS’. 15 ‘Malignant MS’ was defined in 1996 as a ‘disease with a rapid progressive course, leading to significant disability in multiple neurologic systems or death in a relatively short time after disease onset’. 5 However, the authors of this statement did not specify how to quantify any of these criteria, thus leaving the definition somewhat vague. In the following years, they advised that a ‘malignant’ (as well as a ‘benign’) label may be misleading, vary over time and only be determined retrospectively, concluding that such labels should be used with caution. 1
Historical and contemporary attempts at defining aggressive/highly active multiple sclerosis.
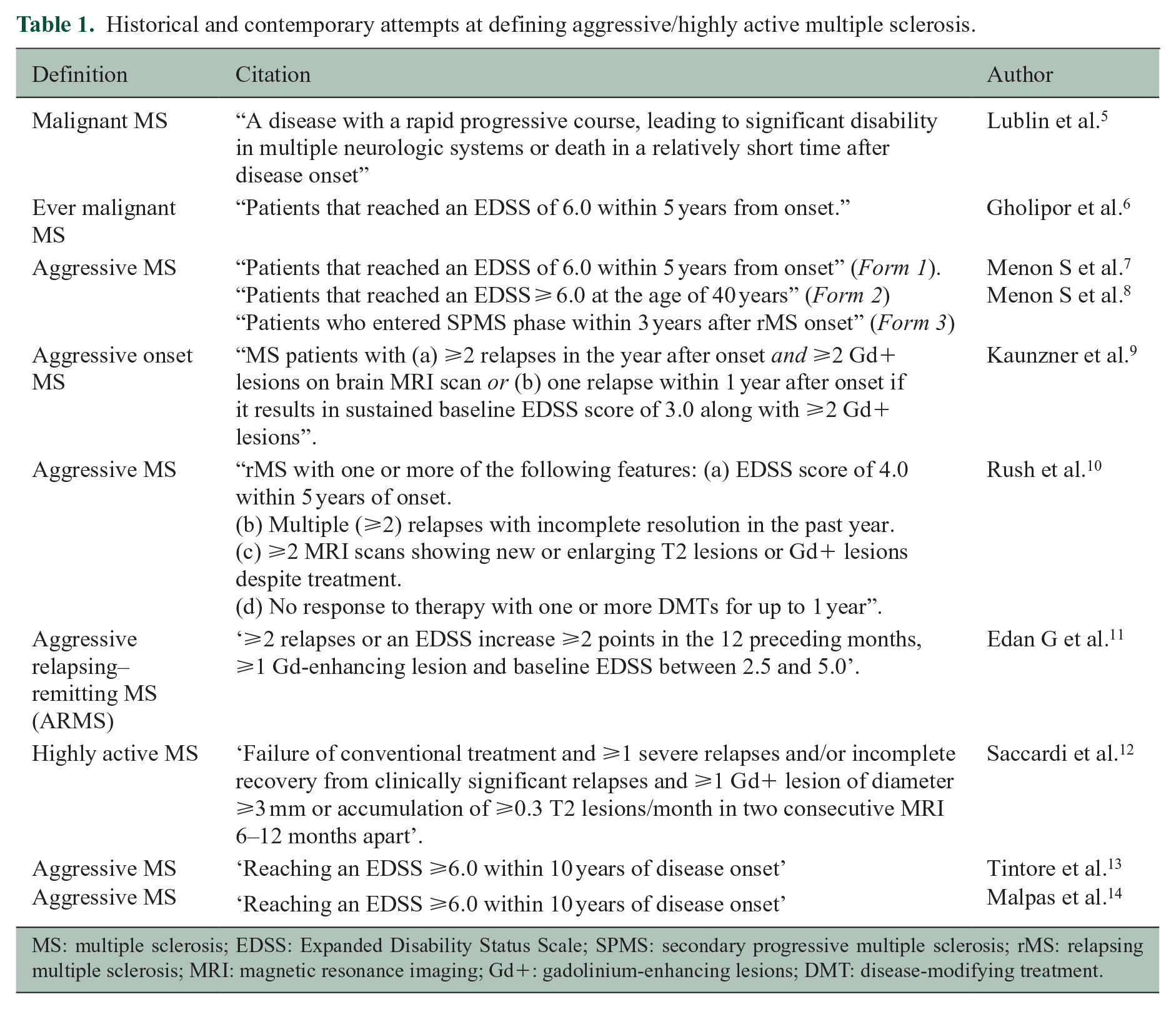
MS: multiple sclerosis; EDSS: Expanded Disability Status Scale; SPMS: secondary progressive multiple sclerosis; rMS: relapsing multiple sclerosis; MRI: magnetic resonance imaging; Gd+: gadolinium-enhancing lesions; DMT: disease-modifying treatment.
The term ‘ever malignant’ was used to define MS patients who reached an Expanded Disability Status Scale (EDSS) of 6.0 within 5 years, which corresponded to 12.1% of a cohort of 478 MS patients. 6 ‘Ever malignant’ cases had significantly more relapses, more motor symptoms and a higher frequency of a progressive onset. 6 In a larger study, 4%–14% of subjects fulfilled the criteria for ‘aggressive MS’.7,8 Here, aggressive MS constituted one of the three forms: form 1 included patients who reached an EDSS of 6 within 5 years, form 2 included those with EDSS ⩾6 at the age of 40 years and form 3 included those who entered SPMS within 3 years after rMS disease onset.7,8 Additional criteria for aggressive onset MS have been described from retrospective assessment as (1) two or more relapses in the year after onset and two or more gadolinium-enhancing (Gd+) lesions on brain magnetic resonance imaging (MRI) or (2) one relapse within a year after onset if it results in sustained baseline EDSS score of 3 along with two or more Gd+ lesions. 9 Based on these criteria, 58 (7.3%) of 783 MS patients were considered to have aggressive onset MS. 9
Other definitions of aggressive MS have been proposed when considering treatment options. One of the rare clinical trials aimed at highly active disease, testing mitoxantrone induction therapy prior to interferon (IFN) β1b, defined aggressive MS as ⩾2 relapses or an EDSS increase ⩾2 points in the 12 preceding months, ⩾1 Gd-enhancing lesion and baseline EDSS between 2.5 and 5.0. 11 Clinicians faced with identifying patients with breakthrough disease eligible for treatment with autologous haematopoetic stem cell transplantation (aHSCT) have characterized such individuals as having ‘highly active MS’, defined as subjects with ‘failure of at least one and up to three DMTs evidenced by ongoing or increased clinical and MRI activity’. 12 A recent review on treatment algorithms for aggressive MS similarly added ‘unresponsiveness to first and/or second line treatment’, as a defining feature to be considered along with clinical and MRI parameters. 10
In spite of these reports with various suggestions for the definition of severe or aggressive MS, there is no consensus about which, if any, of these definitions is most appropriate or most useful in clinical practice or in a research setting. All are hampered by the need for either a retrospective assessment of the disease course or a prolonged prospective evaluation until fulfilling the proposed criteria and thus hinder the ability to make rapid and efficient treatment decisions. Efforts to identify early risk factors for disease severity are ongoing in inception cohorts and registries. Two studies, reported since the conclusion of the workshop, defined aggressive MS as reaching an EDSS ⩾6.0 within 10 years of disease onset. In both cohorts, approximately 5.0–6.0% of patients fulfilled this definition.13,14 Their findings are described in the clinical and MRI subsections in the following.
Characteristics of aggressive MS
Clinical features
Rapid progression of disability is a commonly cited characteristic of aggressive MS. Clinical manifestations that have been associated with an accelerated worsening of disability in MS are summarized in Table 2A, including (a) severe relapse(s) leading to an increase by one EDSS point or greater than or equal to two points in any functional system (FS) and/or the need for hospitalization and/or steroid therapy, (b) multifocal attacks,18,19 (c) incomplete remission20,21 and (d) attacks affecting motor, cognitive, sphincter or cerebellar systems.14,18,22–26 Additional clinical factors are frequent relapses in the first years,16,27,28 short inter-attack interval21,16 and early accrual of disability with superimposed attacks. 16 Severe MS relapses have been more frequently reported among young persons with MS 65 although older age at MS onset is associated with higher risk of disability worsening.24,17 After the Workshop, Malpas et al defined aggressive MS as reaching an EDSS ⩾6.0 within 10 years of disease onset. Indicators of an aggressive disease course included age >35 years at symptom onset, EDSS ⩾3.0 in the first year and presence of pyramidal signs in the first year of disease evolution. Using the MSBase registry, patients with all three features had a 32.0% risk of fulfilling the proposed aggressive MS definition. 14 Some of these findings, such as older age and motor symptoms at disease onset, are in accordance with previous attempts to define aggressive MS in retrospective analyses of prospectively acquired data.6,7
Potential parameters associated with more aggressive disease course (any one or a combination of these characteristics may signal aggressive disease and all are deserving of further exploration and if possible, validation in the context of assessing severity of disease and poor prognosis).

MS: multiple sclerosis; EDSS: Expanded Disability Status Scale; Gd+: gadolinium enhancing lesions; TCR: T-cell receptor; CSF: cerebrospinal fluid; OCB: oligoclonal bands; NfL: neurofilament light chain; CHI3L1: chitinase 3-like 1 protein; MMP9: metallomatrix protein 9.
MRI findings
MRI findings have played a central role in diagnosis, individual evaluation in clinical practice and as a research tool. It can be used to assess disease severity and predict poor prognosis. Several imaging biomarkers have been studied, although not necessarily focusing on aggressive MS (Table 2B, Figure 1). The presence of a high T2 lesion load, >2 Gd + lesions, lesion topography, T1 black holes and early discernible atrophy are well recognized early predictors for disease progression.13,29–34,68–70 A study of over 1000 patients with clinically isolated syndromes (CIS) characteristic of MS showed that a high brain T2 lesion load at baseline was the most robust predictive factor for future disability accrual. 68 Tintore et al. 13 used a Barcelona-based inception cohort and, like Malpas et al., 14 defined aggressive MS as reaching an EDSS ⩾6.0 within 10 years of disease onset. Early radiological biomarkers associated with this outcome were the presence of ⩾20 lesions on T2-weighted images or ⩾2 Gd+ lesions on the brain MRI performed at the time of the first attack. When both MRI conditions were fulfilled, the risk increased to almost 20.0%. 13 The development of >1 Gd+ lesion and new T2 lesions was associated with poorer prognosis at follow-up. 71 In addition, the positive correlation between MRI event-free survival and progression-free survival after aHSCT in MS further supports the predictive value of new MRI lesions for disease progression.72,73
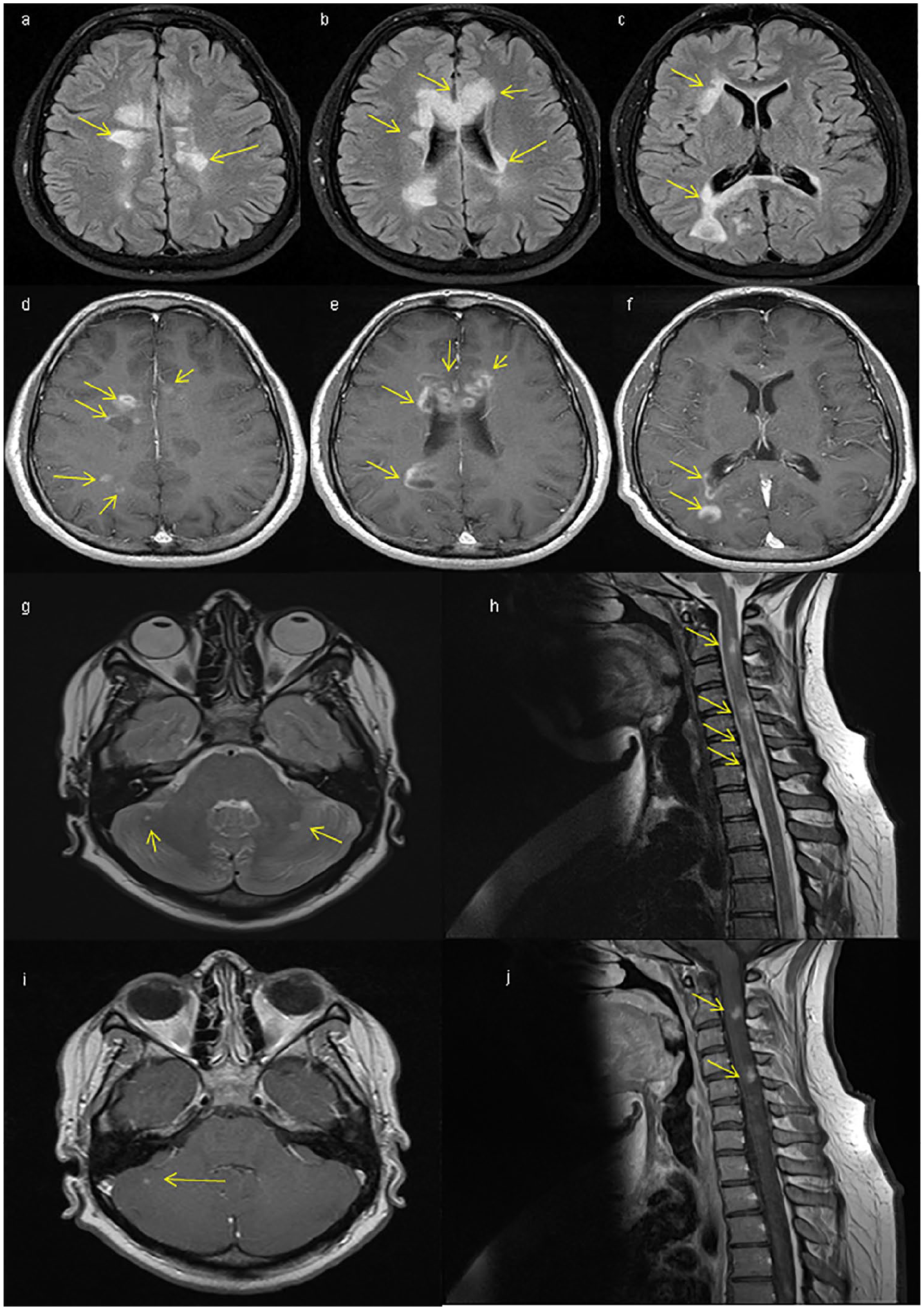
Typical characteristics of aggressive MS on conventional MRI: (a)–(c) T2-FLAIR transverse sequences showing multiple nodular, confluent lesions predominantly affecting the periventricular and callosal topographies (see arrows) in a 33-year-old male patient with a first demyelinating attack. (d)–(f) T1 transverse sequences in the same patient after gadolinium administration show multiple nodular and ring-enhancing lesions. (g) (T2-FLAIR transverse sequence) and (h) (T2 sagittal sequence of the cervical spinal cord) show multiple infratentorial and spinal cord lesions in a 24-year-old female patient with relapsing–remitting multiple sclerosis, several of which showed contrast enhancement despite treatment with (i) and (j) fingolimod.
The presence of ⩾1 spinal cord lesion, symptomatic or asymptomatic, has been reported as a marker of early and long-term disability accrual.30,34 A meta-analysis also showed that spinal cord atrophy significantly correlated with clinical disability. 35 In a study of non-spinal CIS patients, spinal cord factors such as lesion number, change in lesion number and change in upper cervical cord cross-sectional area were independently associated with reaching an EDSS ⩾3.0 at 5 years. Inclusion of brain lesion load and atrophy modestly increased the predictive power of the model, 70 suggesting spinal cord red flags may be more relevant than brain red flags to predict disability, at least as measured by the EDSS. In another study of MS patients, the results suggested that the presence of spinal cord lesions may indicate a higher risk of 6- and 11-year EDSS progression than infratentorial lesions. 74 In addition, the presence of ⩾2 Gd+ lesions and ⩾1 spinal cord lesions at baseline and 1 year and ⩾1 new spinal cord lesions at 3 years of disease evolution were associated with conversion to SPMS at 15 years. 30 Furthermore, the occurrence of cortical lesions and cortical atrophy has been reported as risk factors for early neurological deficit in MS. 37
When assessing tissue loss, the accumulation of hypointense lesions ( ‘black holes’) on T1 sequences correlated with progression rate. 29 As for early brain atrophy, a study in CIS patients showed that a percentage brain volume change (PBVC) decrease below −0.817% in the first year of disease evolution was an independent predictor of a shorter time to a second attack, 75 which could be an indicator of aggressive MS. Evidence of a positive correlation between the disability level and atrophy of the thalamus and basal ganglia has been supported in several studies.76,77 A significant association between deep grey matter brain atrophy rate and time to disability progression was detected in a cohort of more than 1200 MS patients. 36 Interestingly, another study showed that the presence of isolated thalamic atrophy without whole brain atrophy distinguished patients with a higher risk of fast disability progression. 38
More recently, the presence of smouldering lesions has been suggested as a new MRI marker of poor outcome in MS patients since it reflects persistent inflammation, delayed tissue repair and neurodegeneration. 39
Neuropathological aspects of highly active MS
A major contributor to the development of permanent disability in MS is axonal loss, which is an ongoing process from disease onset. 78 This suggests that there should be a strong correlation between rapid disability accumulation and a high rate of axonal damage and loss (Table 2). Key mechanisms resulting in neuronal dysfunction include (1) transection of axons by an inflammatory attack, (2) chronic demyelination causing axonal degeneration, (3) cortical demyelination causing neuritic transection and neuronal death and (4) synaptic spine loss.40,42,43 The observation that macrophage infiltration within active lesions correlates with axonal transection supports the role of inflammatory mechanisms in axonal loss. 40 Axonal loss within chronic demyelinating lesions is thought to be a consequence of loss of myelin trophic support, rendering denuded axons vulnerable to the chronic inflammatory environment with CD8+ cells, microglia and oxidative stress factors. 41
Additional factors contributing to axonal loss occur in the normal appearing white matter (NAWM) involving secondary Wallerian degeneration of injured axons in adjacent white matter plaques. 79 Cortical lesions have been associated with prominent neuronal apoptosis in addition to axonal and dendritic transection, and the presence of such lesions has been suggested as a potential biomarker for risk of early disability development. 80
Taken together, extensive widespread neuroaxonal damage and loss in MS may be viewed as the neuropathologic counterpart to clinically aggressive MS, but this needs to be validated in clinicopathological correlated studies. In clinical practice, ongoing axonal loss may be the underlying pathology in patients with sustained progression despite treatment with one or more DMTs.
Peripheral immune correlates of highly active MS
Alterations in the peripheral immune system associated with highly active MS have been poorly studied. However, the observation of sustained suppression of active neuroinflammation following aHSCT in patients with aggressive/highly active MS has provided insights into immune mechanisms that might be associated with a highly inflammatory clinical phenotype in MS 81 (Table 2D). A significant shift in the balance between regulatory and proinflammatory cells in the circulation has been detected in patients with active MS that underwent successful aHSCT.44–47 Proportionally increased levels of CD4 + Foxp3 + regulatory T-cells (Tregs), memory CD4 + and CD8 + cells, CD56high natural killer (NK) cells and diminished Th17 cell responses have been observed during peripheral immune reconstitution after aHSCT in MS.44–47 In addition, successful outcomes after aHSCT in MS patients with ‘a poor prognosis’ correlated with more diverse T-cell receptor (TCR) repertoires of CD4 + cells suggesting that low TCR diversity may be a risk factor for a highly inflammatory disease course. 49 Altogether, these findings indicate a possible association between dysregulated subgroups of T cells and NK cells with highly active MS. Such data, not specifically gathered to define aggressive MS, deserve further exploration and validation as potential immune markers for the disease.
CSF and blood biomarkers with prognostic values (Table 2E and Table 3)
Intrathecal IgG synthesis at MS onset was associated with increased risk of disability worsening within 4 years although intrathecal IgG synthesis is present in 60% of MS patients independent of disease severity or prognosis. 48 Furthermore, the presence of IgM OCB correlated with higher disease activity, EDSS progression and brain atrophy in rMS.50,51
A selection of biomarkers in CNS and periphery and their association with prognosis.
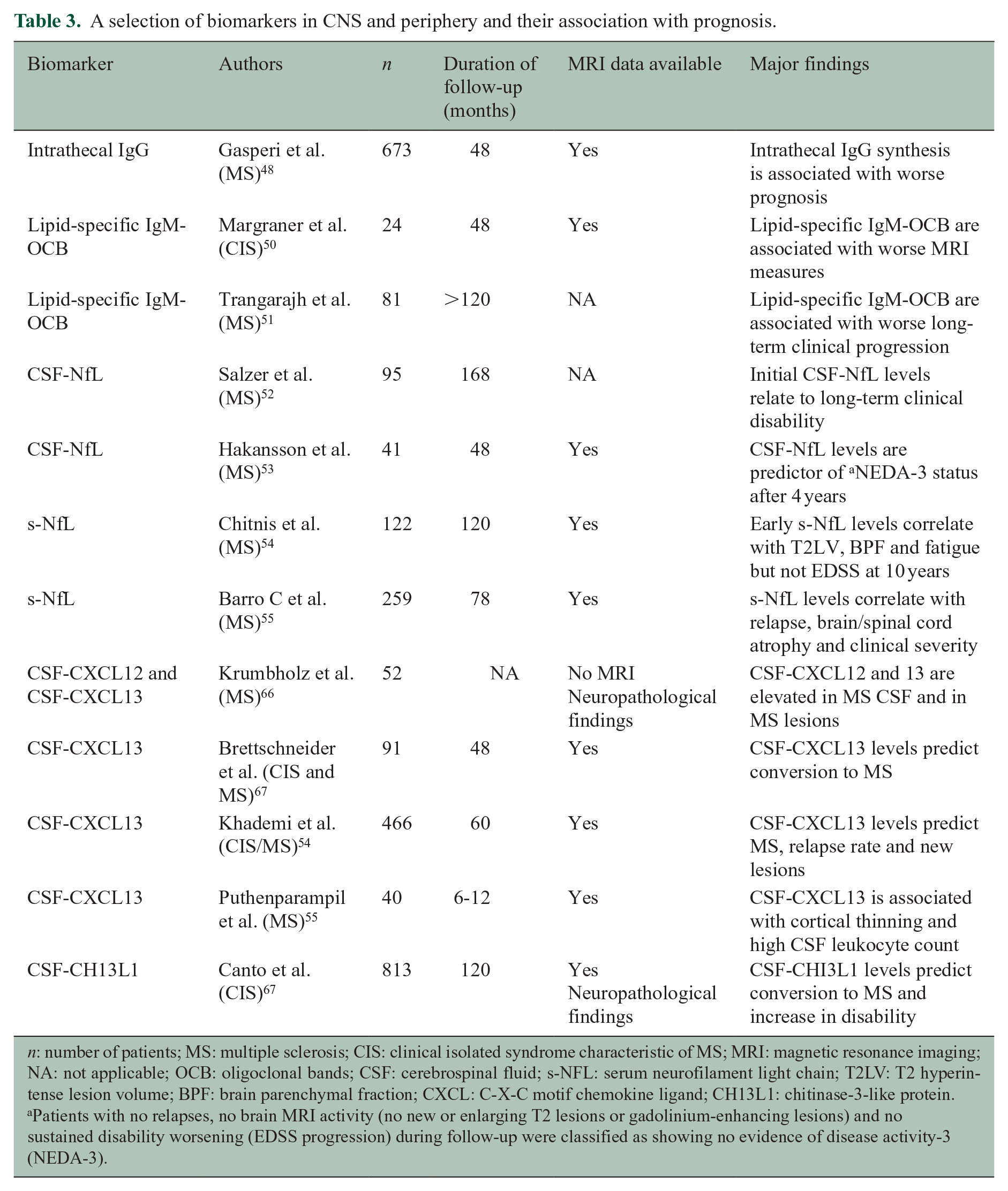
n: number of patients; MS: multiple sclerosis; CIS: clinical isolated syndrome characteristic of MS; MRI: magnetic resonance imaging; NA: not applicable; OCB: oligoclonal bands; CSF: cerebrospinal fluid; s-NFL: serum neurofilament light chain; T2LV: T2 hyperintense lesion volume; BPF: brain parenchymal fraction; CXCL: C-X-C motif chemokine ligand; CH13L1: chitinase-3-like protein.
Patients with no relapses, no brain MRI activity (no new or enlarging T2 lesions or gadolinium-enhancing lesions) and no sustained disability worsening (EDSS progression) during follow-up were classified as showing no evidence of disease activity-3 (NEDA-3).
Evaluation of neurofilament light chain (NfL) levels in both CSF (cNfL) and serum (sNfL) has shown strong promise as a marker for disease activity and disability correlated with neuroaxonal damage in MS and has been implemented as part of routine clinical assessment in some countries. To date, however, a correlation of NfL levels with disease severity has not been studied. Levels of cNfL were associated with both new T2 lesions and brain volume loss during follow-up; cNfL at MS diagnosis correlated with Multiple Sclerosis Severity Scale (MSSS) estimates during a follow-up period of 8–20 years.52,53 In a related finding, sNfL levels correlated with risk of future relapses, 53 disability worsening and brain T2 lesion load.54,55 Increased sNfL was also found to be a negative predictor for MS, correlating with brain atrophy and disability development in early MS.54,55 Some studies have attempted to establish predictive cut-off values of NfL levels. Patients with cNfL levels >386 ng/L had a higher risk of conversion to SPMS than patients with lower levels, 52 although this cut-off has not been fully validated. Another study assessed stratified cNfl and sNfL measurements in rMS patients during the first years of participation in a clinical trial on IFN-β versus placebo. Upper tertile year 2 cNfl and year 3 sNfL levels were predictive of reaching an EDSS ⩾6.0 at year 8 from inclusion compared to lower tertile levels. Similarly, upper tertile year 4 sNfL concentration was predictive of reaching an EDSS score ⩾6.0 at 15 years, with NfL concentrations being complementary to 2-year brain parenchymal fraction change in predicting long-term outcomes. 82
Evaluation of C-X-C motif chemokine ligand 13 (CXCL13), also called B-cell attracting chemokine 1 (BCA-1), has recently been used to assess inflammatory activity in MS. The chemokine CXCL13 is considered to reflect B-cell activity in the CNS, and increased CSF CXCL13 levels were found to correlate with conversion from CIS to clinically definite MS and with relapse rates.56,66,67 Furthermore, CSF CXCL13 levels were associated with cortical atrophy in MS suggesting its value as a marker to predict disability progression. 57
Since presence of cortical lesions may be considered as a risk factor for early MS disability development, 32 there have been attempts to develop surrogate non-imaging biomarkers for assessment of cortical damage. Increased levels of a specific panel of proinflammatory cytokines and chemokines including TNF, sTNFR1, IFN γ, CXCL12, CXCL13, IL6, IL8, IL10, BAFF, APRIL, LIGHT, TWEAK, MMP-2, pentraxin3 and sCD163 distinguished patients with high cortical lesion load at diagnosis compared to patients with low cortical lesion load. 83
Other potential CSF biomarkers that reflect inflammatory activity and may have prognostic value include proteolytic enzyme matrix metalloproteinase-9 (MMP9), osteopontin and chitinase 3-like 1 protein (CHI3L1). MMP9 is considered to have a central role in MS pathogenesis with functions relating to CNS tissue destruction. The finding that progressive MS with inflammatory activity had increased levels of CSF MMP9 suggests its potential as a marker to distinguish progressive patients that could benefit from anti-inflammatory therapies. 59 CHI3L1 correlated with disease severity in several inflammatory diseases and was an independent risk factor for development of EDSS 3 in a multivariate study on more than 800 European CIS patients. 58 Importantly, none of these biomarkers have yet been established as reliable markers to predict aggressive MS and should currently only be viewed as indicators with potential prognostic values and as a fertile area for research and validation.
Genetic markers of MS disease activity
MS risk genes have been investigated for their influence on MS disease outcome but the results have been conflicting63,84 (Table 2F). The most dominant MS risk gene, HLA-DRB1*15:01, correlated with development of increased number of T2 MRI lesions and loss of brain volume in MS. 60 A German study demonstrated that PD-1 polymorphisms were associated with a progressive disease course, which was considered to be a consequence of impaired PD-1-mediated inhibition of T cell activation. 61 A genome-wide association study detected variants of the glycosylation enzyme MGAT5 that influenced the severity of MS 62 and carriers of the APOE-epsilon 4 allele had a more severe disease progression. 63
Non-immune-related genes such as those involved in neuroprotective or regenerative mechanisms have emerged as possible risk factors for MS disease outcome heterogeneity.85,86 Genetically determined axonal response to inflammation and environmental factors might be associated with clinical outcome and disability progression in MS. An intriguing example was the detection that allelic variants of the neuroprotective ciliary neurotrophic factor (CNTF) were associated with disease onset, course and severity of experimental autoimmune encephalomyelitis (EAE) in mice. 87 Furthermore, polymorphisms of the brain-derived neurotrophic factor (BDNF) have in some studies been shown to impact on grey matter tissue damage in MS. 64 However, current data on gene variants associated with aggressive MS are very sparse and controversial; additional genetic studies may determine whether the clinical cohort of aggressive MS can be genetically distinguished from other disease phenotypes.
Achievements, gaps in knowledge and future perspectives
Clinical, paraclinical and laboratory assessments all have the potential to be used in categorizing MS patients with a prognosis of poor outcomes over a short period of time, although most have not been evaluated with this goal in mind. Disease severity and activity are essential parameters when considering the right treatment for patients. 88 While guidelines exist for treatment of rMS, PPMS and SPMS, specific guidance for how to approach treatment for severe, rapidly aggressive MS are lacking. 88 To develop such guidelines, and to better manage those patients with aggressive disease, it is important to clearly identify subjects with initial or evolving signs of aggressive disease course as early as possible and offer them a highly efficient treatment as soon as possible after disease onset to mitigate breakthrough disease and progression of permanent disability.
Based on the discussion held during the ECTRIMS workshop on aggressive MS and on recent results from retrospective studies with prospectively collected data, we believe that several identified clinical and imaging features, possibly along with elevated CSF and/or serum NfL levels, might be considered as the most promising characteristics of highly active MS (Table 4).
‘Red flags’ that may indicate highly active MS.

MS: multiple sclerosis; DMT: disease modifying treatment; EDSS: Expanded Disability Status Scale; Gd +: gadolinium enhancing; PBVC: percentage brain volume change; NfL: neurofilament light chain; CSF: cerebrospinal fluid.
Includes relapsing and progressive forms of MS.
A limitation of this proposal is that several of these features were not studied in the context of aggressive MS or weighted against other suspected risk factors. The identification of several risk factors, however, depended on establishing a working definition of aggressive MS which, in two studies, was reaching an EDSS ⩾6.0 within 10 years of disease onset. Clinical risk factors of individuals who might be considered to have aggressive MS are age >35 years at symptom onset, EDSS ⩾3.0 in the first year and presence of pyramidal signs in the first year of disease evolution. 14 Older age and motor symptoms at disease onset were also identified as risk factors in previous attempts to define aggressive MS.6,7 MRI risk factors include the presence of ⩾20 T2 lesions or ⩾2 Gd+ lesions on the brain MRI performed at disease onset. 13 When present, these factors may serve as ‘red flags’ to define a patient as a candidate for early, more aggressive therapeutic intervention. This suggestion must be considered a working definition that needs to be solidified through future research, requiring validation in other cohorts to confirm or refute their short- and long-term value in the clinical practice. Conversely, we also have to acknowledge that several parameters potentially associated with aggressive MS (Table 2) may be useful only for research purposes to understand this phenotype (i.e. neuroaxonal damage). In this sense, it will be important to identify those with translational potential.
As noted in the manuscript, the list of current ‘red flags’ for aggressive disease is far from complete. Notably missing are immunological assays, advanced imaging, pathological, genetic, and many potential body fluid biomarkers mentioned above. Identifying their differential contribution of relapse activity and disease progression is difficult, as disability may occur due to the consequences of inflammation and/or pure neurodegenerative mechanisms. Rather, it is important to study the degree to which inflammation and neurodegeneration factors (and their possible interactions) are involved in the etiopathogenesis of aggressive MS. This may aid in defining not only aggressive MS in relapsing forms but also in progressive phenotypes in which available evidence is even scarcer.
Importantly, whereas recent studies are based on findings at disease onset or during the first year of the disease,13,14 potential risk factors should be assessed throughout the disease evolution. MS may become highly active after years of stability in some patients, and persons with a high functional reserve may take longer to develop disability. 89
In addition, as the proportion of patients with characteristics of aggressive MS is low, collaborative studies based on large observational registries and focused cohorts are necessary. These studies enable prognostication that is both generalizable and comprehensive. In addition, robust analytical approaches of the most relevant risk factors, like Bayesian analyses, are a requisite of a shift from studies of associations at the group level towards individual prognosis. Afterwards, replication of novel associations in independent validation cohorts is essential.
While objective markers for MS disease severity are desirable, one cannot ignore the value of clinical judgement, independent of the presence or absence of markers, especially in the context of patients being followed over time by the same physician.
Supplemental Material
MSJ925369_supplemental_table_1 – Supplemental material for Aggressive multiple sclerosis (1): Towards a definition of the phenotype
Supplemental material, MSJ925369_supplemental_table_1 for Aggressive multiple sclerosis (1): Towards a definition of the phenotype by Ellen Iacobaeus, Georgina Arrambide, Maria Pia Amato, Tobias Derfuss, Sandra Vukusic, Bernhard Hemmer, Mar Tintore and Lou Brundin in Multiple Sclerosis Journal
Footnotes
Acknowledgements
The authors wish to thank ECTRIMS for financial and administrative support of the focused workshop that served as the basis for this paper and all workshop attendees (Supplemental Table 2) for their participation. All attendees had the opportunity to review and comment upon an advanced draft of the manuscript prior to submission. We also wish to thank Dr Stephen C. Reingold for assistance with manuscript writing and editing, supported by ECTRIMS.
Declaration of Conflicting Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship and/or publication of this article: E.I. has honoraria for advisory boards or lecturing for Sanofi Genzyme and Merck outside the submitted work. G.A. has received compensation for consulting services or participation in advisory boards from Sanofi, Merck and Roche; research support from Novartis; travel expenses for scientific meetings from Novartis, Roche and Stendhal; and speaking honoraria from Sanofi and Merck. M.P.A. has received research grants and personal fees as a speaker and member of advisory boards by Biogen, Sanofi Genzyme, Merck Serono, Novartis, Roche and Teva, outside the submitted work. T.D. reports grants from Novartis and Biogen. Dr Derfuss’ institution received financial support from Novartis, Biogen, Roche, Merck Serono, Celgene, Sanofi Genzyme, MedDay, GeNeuro, Mitsubishi Pharma and Actelion for his activities as steering committee or advisory board member or consultant. S.V. has received grants, personal fees and non-financial support from Biogen, Celgene, Geneuro, Genzyme, Medday, Merck Serono, Novartis, Roche, Sanofi and Teva, outside the submitted work. B.H. has served on scientific advisory boards for Novartis and as DMSC member for AllergyCare and TG therapeutics; he or his institution have received speaker honoraria from Desitin; holds part of two patents; one for the detection of antibodies against KIR4.1 in a subpopulation of MS patients and one for genetic determinants of neutralizing antibodies to interferon. M.T. has received compensation for consulting services and speaking honoraria from Almirall, Bayer Schering Pharma, Biogen-Idec, Genzyme, Merck Serono, Novartis, Roche, Sanofi-Aventis and Teva Pharmaceuticals. M.T. is co-editor of Multiple Sclerosis Journal ETC. L.B. has received honoraria for advisory boards for Biogen, Sanofi-Genzyme, Novartis and Teva and has received lecturing fees from Biogen, Novartis, Teva and Sanofi-Genzyme outside the submitted work.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship and/or publication of this article: The workshop on which the manuscript is based was supported in its entirety by the European Committee on Treatment and Research in Multiple Sclerosis (ECTRIMS).
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.