
Abstract
The natural history of multiple sclerosis (MS) is highly heterogeneous. A subgroup of patients has what might be termed aggressive MS. These patients may have frequent, severe relapses with incomplete recovery and are at risk of developing greater and permanent disability at the earlier stages of the disease. Their therapeutic window of opportunity may be narrow, and while it is generally considered that they will benefit from starting early with a highly efficacious treatment, a unified definition of aggressive MS does not exist and data on its treatment are largely lacking. Based on discussions at an international focused workshop sponsored by the European Committee for Treatment and Research in Multiple Sclerosis (ECTRIMS), we review our current knowledge about treatment of individuals with aggressive MS. We analyse the available evidence, identify gaps in knowledge and suggest future research needed to fill those gaps. A companion paper details the difficulties in developing a consensus about what defines aggressive MS.
Introduction
Over the past 25 years, more than a dozen therapies have been approved by regulators for use in MS. 1 Safety and efficacy have been demonstrated through prospective randomized controlled clinical trials, most often including subjects with relapsing forms of the disease, relatively early in the disease process, and with demonstrated inflammatory underlying disease biology.2,3 Historically, such patients have been deemed to be a subgroup that most likely benefits from treatment, and thus trial populations have been enriched to focus on these individuals. Only recently have clinical trials been successful in subjects with progressive forms of MS (pMS).4,5
A long-recognized subgroup of patients has what might be termed aggressive MS. An aggressive disease course may manifest from onset or develop during the disease course. These patients may have frequent, severe relapses with incomplete recovery and are at risk of developing greater and permanent disability within a short time frame.6,7 Although it was not possible to come to consensus about a definition of aggressive MS at the end of the workshop, we considered radiological, clinical and biological features that might be used to obtain such a consensus. Two recent papers proposed reaching an Expanded Disability Status Scale (EDSS) ⩾ 6.0 within 10 years of disease onset as aggressive MS (please refer to the companion paper 7 for a more detailed discussion).8,9 These and several other definitions are based on relapse frequency or severity, lack of recovery, disability accrual or magnetic resonance imaging (MRI) lesions.
While it is tempting to extrapolate results from past successful clinical trials to all patients with MS when considering treatment options, data are not necessarily applicable to individuals with a more aggressive disease because of differences in underlying disease processes, 10 demographic considerations that may have an impact on treatment outcomes, 6 differences in comorbidities 11 and other factors compared with the general relapsing MS trial population.
While prospective randomized clinical trial data on treatment of enriched populations of aggressive MS are largely lacking, suggestive evidence is available from other types of studies, including data from other relapsing inflammatory demyelinating diseases (Table 1) and data from subgroups of patients in MS clinical trials using a variety of descriptions of what is considered aggressive disease (Tables 2–4). Here, we present what is known about treatment of more aggressive forms of MS and provide suggestions for future research to fill gaps in knowledge about the best therapeutic management of these patients.
Treatment of severe relapse: types of patients included and definitions of relapse severity.
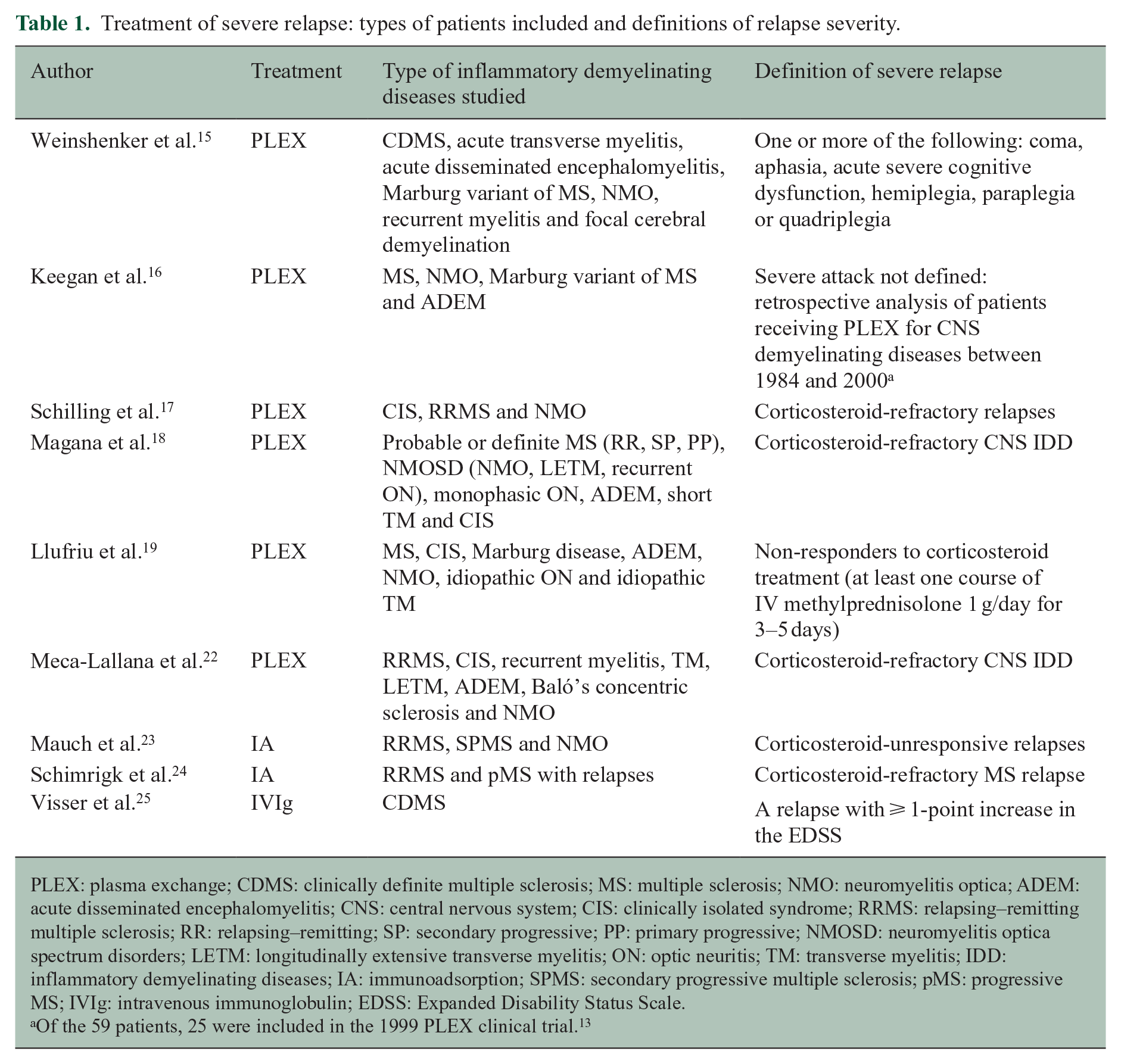
PLEX: plasma exchange; CDMS: clinically definite multiple sclerosis; MS: multiple sclerosis; NMO: neuromyelitis optica; ADEM: acute disseminated encephalomyelitis; CNS: central nervous system; CIS: clinically isolated syndrome; RRMS: relapsing–remitting multiple sclerosis; RR: relapsing–remitting; SP: secondary progressive; PP: primary progressive; NMOSD: neuromyelitis optica spectrum disorders; LETM: longitudinally extensive transverse myelitis; ON: optic neuritis; TM: transverse myelitis; IDD: inflammatory demyelinating diseases; IA: immunoadsorption; SPMS: secondary progressive multiple sclerosis; pMS: progressive MS; IVIg: intravenous immunoglobulin; EDSS: Expanded Disability Status Scale.
Of the 59 patients, 25 were included in the 1999 PLEX clinical trial. 13
Studies investigating the use of oral DMTs for the treatment of aggressive MS. a .
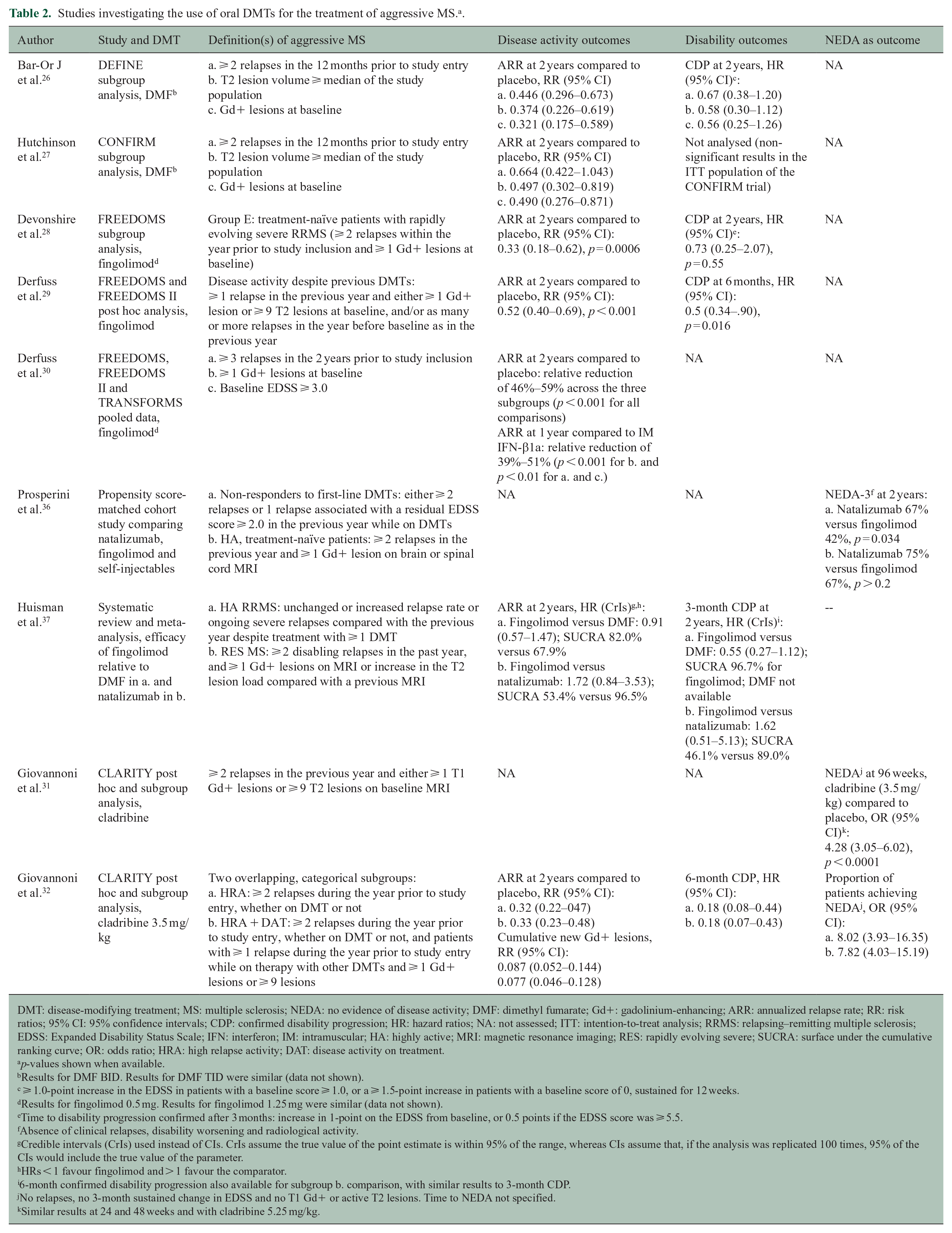
DMT: disease-modifying treatment; MS: multiple sclerosis; NEDA: no evidence of disease activity; DMF: dimethyl fumarate; Gd+: gadolinium-enhancing; ARR: annualized relapse rate; RR: risk ratios; 95% CI: 95% confidence intervals; CDP: confirmed disability progression; HR: hazard ratios; NA: not assessed; ITT: intention-to-treat analysis; RRMS: relapsing–remitting multiple sclerosis; EDSS: Expanded Disability Status Scale; IFN: interferon; IM: intramuscular; HA: highly active; MRI: magnetic resonance imaging; RES: rapidly evolving severe; SUCRA: surface under the cumulative ranking curve; OR: odds ratio; HRA: high relapse activity; DAT: disease activity on treatment.
p-values shown when available.
Results for DMF BID. Results for DMF TID were similar (data not shown).
⩾ 1.0-point increase in the EDSS in patients with a baseline score ⩾ 1.0, or a ⩾ 1.5-point increase in patients with a baseline score of 0, sustained for 12 weeks.
Results for fingolimod 0.5 mg. Results for fingolimod 1.25 mg were similar (data not shown).
Time to disability progression confirmed after 3 months: increase in 1-point on the EDSS from baseline, or 0.5 points if the EDSS score was ⩾ 5.5.
Absence of clinical relapses, disability worsening and radiological activity.
Credible intervals (CrIs) used instead of CIs. CrIs assume the true value of the point estimate is within 95% of the range, whereas CIs assume that, if the analysis was replicated 100 times, 95% of the CIs would include the true value of the parameter.
HRs < 1 favour fingolimod and > 1 favour the comparator.
6-month confirmed disability progression also available for subgroup b. comparison, with similar results to 3-month CDP.
No relapses, no 3-month sustained change in EDSS and no T1 Gd+ or active T2 lesions. Time to NEDA not specified.
Similar results at 24 and 48 weeks and with cladribine 5.25 mg/kg.
Studies investigating the use of monoclonal antibodies for the treatment of aggressive MS.
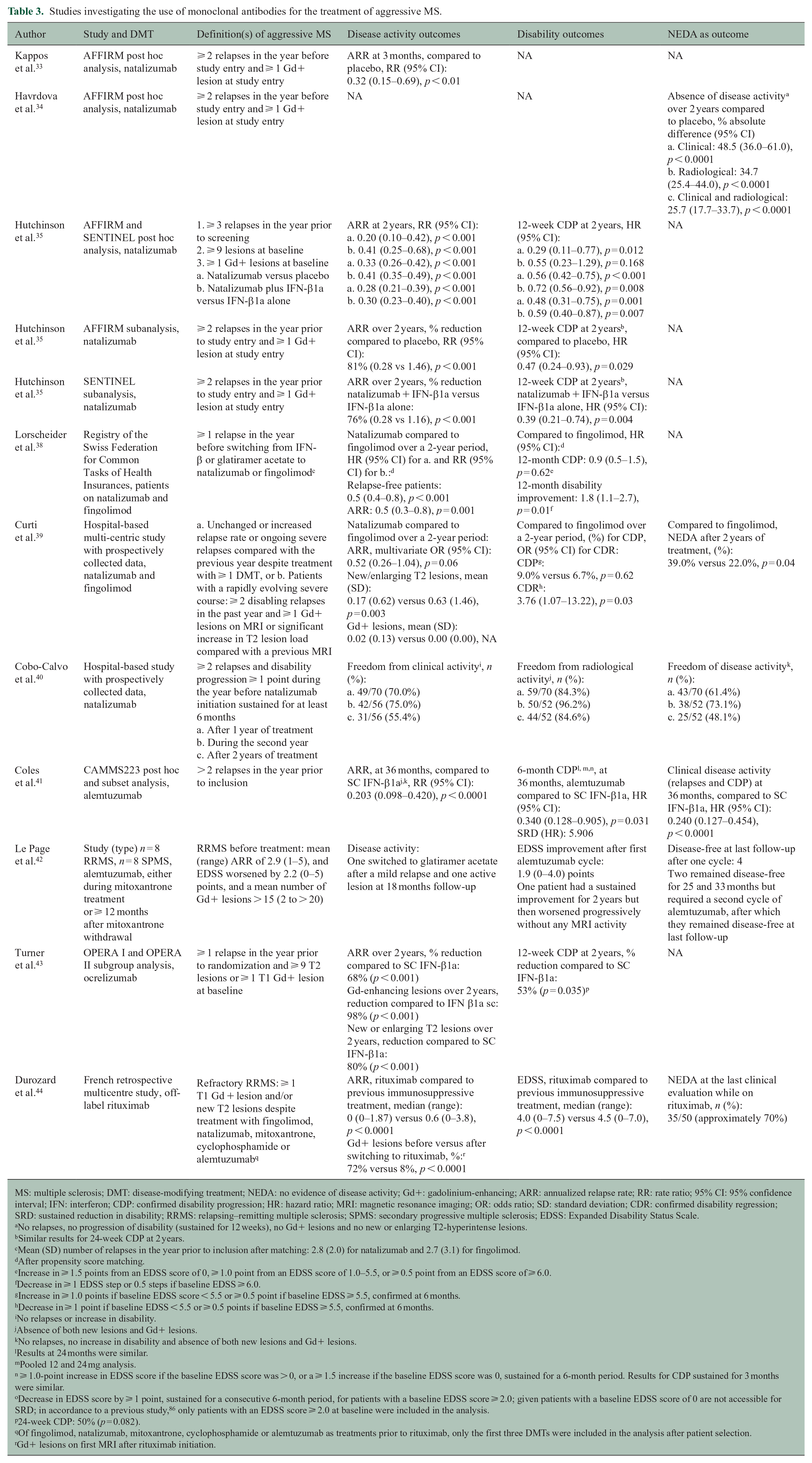
MS: multiple sclerosis; DMT: disease-modifying treatment; NEDA: no evidence of disease activity; Gd+: gadolinium-enhancing; ARR: annualized relapse rate; RR: rate ratio; 95% CI: 95% confidence interval; IFN: interferon; CDP: confirmed disability progression; HR: hazard ratio; MRI: magnetic resonance imaging; OR: odds ratio; SD: standard deviation; CDR: confirmed disability regression; SRD: sustained reduction in disability; RRMS: relapsing–remitting multiple sclerosis; SPMS: secondary progressive multiple sclerosis; EDSS: Expanded Disability Status Scale.
No relapses, no progression of disability (sustained for 12 weeks), no Gd+ lesions and no new or enlarging T2-hyperintense lesions.
Similar results for 24-week CDP at 2 years.
Mean (SD) number of relapses in the year prior to inclusion after matching: 2.8 (2.0) for natalizumab and 2.7 (3.1) for fingolimod.
After propensity score matching.
Increase in ⩾ 1.5 points from an EDSS score of 0, ⩾ 1.0 point from an EDSS score of 1.0–5.5, or ⩾ 0.5 point from an EDSS score of ⩾ 6.0.
Decrease in ⩾ 1 EDSS step or 0.5 steps if baseline EDSS ⩾ 6.0.
Increase in ⩾ 1.0 points if baseline EDSS score < 5.5 or ⩾ 0.5 point if baseline EDSS ⩾ 5.5, confirmed at 6 months.
Decrease in ⩾ 1 point if baseline EDSS < 5.5 or ⩾ 0.5 points if baseline EDSS ⩾ 5.5, confirmed at 6 months.
No relapses or increase in disability.
Absence of both new lesions and Gd+ lesions.
No relapses, no increase in disability and absence of both new lesions and Gd+ lesions.
Results at 24 months were similar.
Pooled 12 and 24 mg analysis.
⩾ 1.0-point increase in EDSS score if the baseline EDSS score was > 0, or a ⩾ 1.5 increase if the baseline EDSS score was 0, sustained for a 6-month period. Results for CDP sustained for 3 months were similar.
Decrease in EDSS score by ⩾ 1 point, sustained for a consecutive 6-month period, for patients with a baseline EDSS score ⩾ 2.0; given patients with a baseline EDSS score of 0 are not accessible for SRD; in accordance to a previous study, 86 only patients with an EDSS score ⩾ 2.0 at baseline were included in the analysis.
24-week CDP: 50% (p = 0.082).
Of fingolimod, natalizumab, mitoxantrone, cyclophosphamide or alemtuzumab as treatments prior to rituximab, only the first three DMTs were included in the analysis after patient selection.
Gd+ lesions on first MRI after rituximab initiation.
Studies investigating the use of aHSCT in the treatment of aggressive MS.
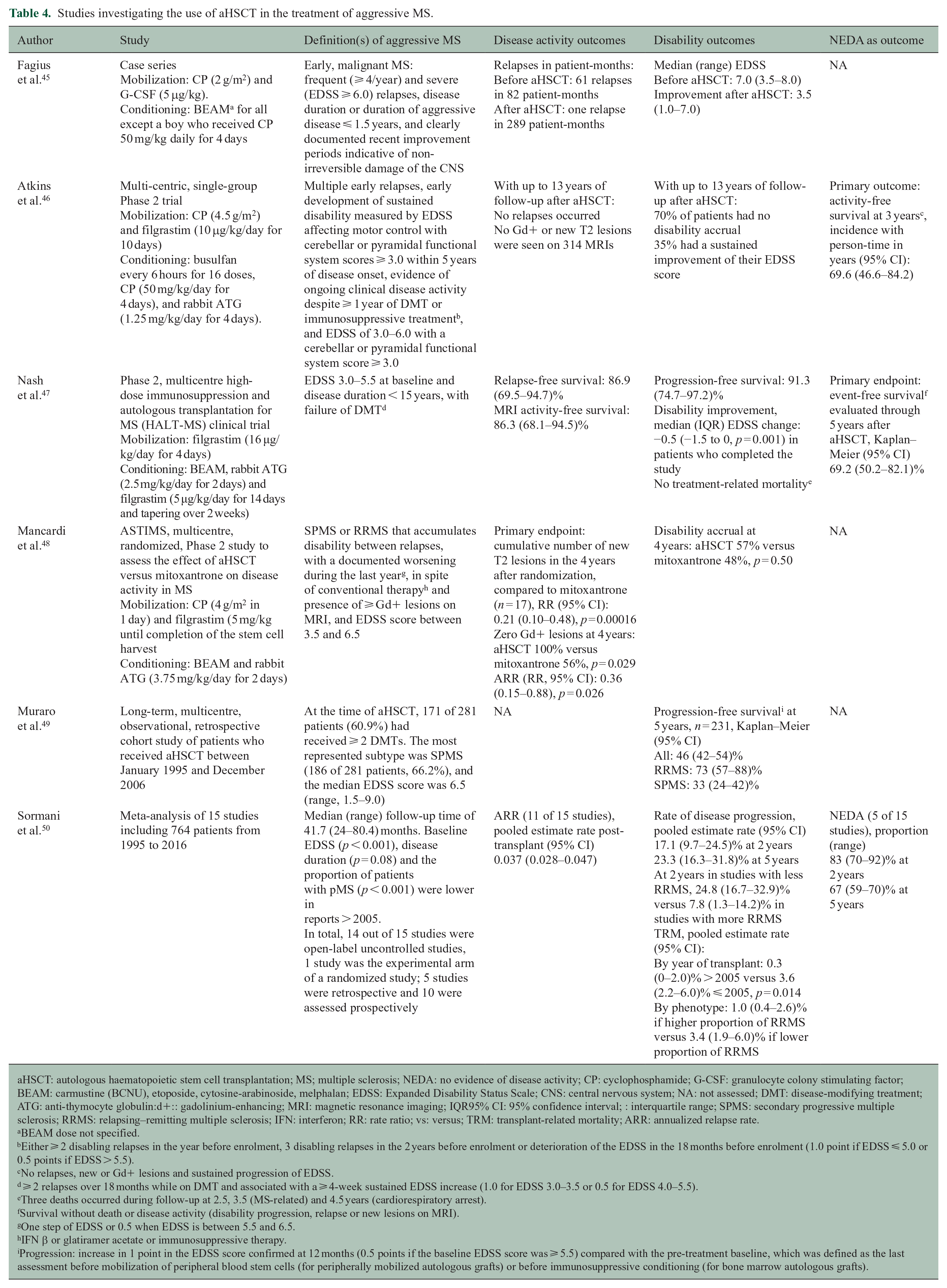
aHSCT: autologous haematopoietic stem cell transplantation; MS; multiple sclerosis; NEDA: no evidence of disease activity; CP: cyclophosphamide; G-CSF: granulocyte colony stimulating factor; BEAM: carmustine (BCNU), etoposide, cytosine-arabinoside, melphalan; EDSS: Expanded Disability Status Scale; CNS: central nervous system; NA: not assessed; DMT: disease-modifying treatment; ATG: anti-thymocyte globulin:d+:: gadolinium-enhancing; MRI: magnetic resonance imaging; IQR95% CI: 95% confidence interval; : interquartile range; SPMS: secondary progressive multiple sclerosis; RRMS: relapsing–remitting multiple sclerosis; IFN: interferon; RR: rate ratio; vs: versus; TRM: transplant-related mortality; ARR: annualized relapse rate.
BEAM dose not specified.
Either ⩾ 2 disabling relapses in the year before enrolment, 3 disabling relapses in the 2 years before enrolment or deterioration of the EDSS in the 18 months before enrolment (1.0 point if EDSS ⩽ 5.0 or 0.5 points if EDSS > 5.5).
No relapses, new or Gd+ lesions and sustained progression of EDSS.
⩾ 2 relapses over 18 months while on DMT and associated with a ⩾ 4-week sustained EDSS increase (1.0 for EDSS 3.0–3.5 or 0.5 for EDSS 4.0–5.5).
Three deaths occurred during follow-up at 2.5, 3.5 (MS-related) and 4.5 years (cardiorespiratory arrest).
Survival without death or disease activity (disability progression, relapse or new lesions on MRI).
One step of EDSS or 0.5 when EDSS is between 5.5 and 6.5.
IFN β or glatiramer acetate or immunosuppressive therapy.
Progression: increase in 1 point in the EDSS score confirmed at 12 months (0.5 points if the baseline EDSS score was ⩾ 5.5) compared with the pre-treatment baseline, which was defined as the last assessment before mobilization of peripheral blood stem cells (for peripherally mobilized autologous grafts) or before immunosuppressive conditioning (for bone marrow autologous grafts).
Materials and methods
Aggressive MS, its definition and management were discussed at the 1–2 March 2018 Focused Workshop on Aggressive Multiple Sclerosis held in Brussels, Belgium, supported by the European Committee for Treatment and Research in Multiple Sclerosis (ECTRIMS). A review of the pathology, demographic and clinical characteristics, and prognosis of aggressive MS, as well as the methods used to try to reach a consensus definition are presented in a companion article. 7 Prior to the workshop, specific treatments or groups of treatments were assigned to different participating MS specialists who conducted a relevant literature search on PubMed including but not limited to the terms: ‘treatment x’ AND ‘multiple sclerosis’ OR ‘MS’ AND ‘aggressive’ OR ‘highly active’. The search was performed up to March 2018 with no limits on language. At the workshop, attendees (Supplemental Table 1) presented and discussed available evidence that could inform treatment options for their assigned subgroup of patients based on data derived from post hoc analyses, subgroup analyses, meta-analyses, registries, cohorts and the scarce treatment trials designed especially for aggressive MS. Subsequent to the workshop, the discussions were summarized and the references were updated using a similar literature search strategy. Gaps in knowledge were identified, and strategies for future research to better understand treatment of aggressive MS were discussed.
Results
Acute treatment
Relapses, including frequency, duration and severity, are clinical indicators of MS disease activity. While usually self-limiting, some relapses can be disabling and recovery can be incomplete, resulting in long-term accumulating disability. 12 Clinical trials of agents for treating relapsing forms of MS (rMS) have examined the impact of treatment on relapse rate, but few have examined the impact on shortening the duration or reducing the severity of a relapse. Trials assessing relapse treatments often have heterogeneous inclusion criteria, including patients with differing MS phenotypes and sometimes different demyelinating diseases (Table 1).
High-dose corticosteroids
High-dose intravenous (IV) or oral corticosteroids are the first-line treatment for relapses in MS.13,14 Corticosteroids are believed to have an impact on MS relapses by exerting a rapid immunosuppressant effect. 11 Data indicate that IV or oral corticosteroids decrease the severity of relapses as measured by the EDSS or the Kurtzke Functional System Scale and speed patient recovery in rMS,12–14 but evidence of its effect on relapses in pMS is very limited. 13
Plasmapheresis and immunoadsorption
Plasma exchange (PLEX) has been used for patients with persistently severe neurological deficits, although definitions of severity vary among studies. 12 Evidence of usefulness of PLEX in MS arises largely from a randomized, sham-controlled, double-masked trial in patients with severe attacks of inflammatory demyelinating diseases (not all with MS) who failed to recover after IV high-dose methylprednisolone treatment. 15 Moderate or greater improvement occurred in 42.1% of courses of active treatment with PLEX compared to 5.9% of courses of sham treatment (p = 0.011). Initiation of treatment within the first 20 days of an attack was one of the factors associated with moderate or marked improvement, and a beneficial effect was also observed with treatment within the first 20–60 days in some cases. 16 In general, clinical improvement was seen after three PLEX treatments,17,18 and continuing improvement was observed as late as 6 months after the completion of PLEX in treatment responders.15,16,18,19 Only anecdotal evidence exists for a role of PLEX in corticosteroid-refractory superimposed relapses in secondary progressive MS (SPMS).20,21
MRI predictors of response to PLEX include the presence of gadolinium-enhancing (Gd+) lesions and lesions with mass effect, according to results from an exploratory analysis of 153 patients with central nervous system inflammatory demyelinating diseases. 18 Conversely, a smaller observational, pilot study, including 15 patients with different demyelinating diseases, showed no significant association between the degree of resolution of the radiological activity and the clinical response to PLEX. 22
In contrast to PLEX, immunoadsorption (IA) removes antibodies and immune complexes while avoiding plasma product substitution.23,24 Similarly to PLEX, results from retrospective studies suggest that IA might be most effective after three such procedures in MS patients with corticosteroid-refractory relapses. 23 Moderate to marked improvement from attack-related disability as measured by the EDSS can be observed in approximately 70% of cases, including a subset of patients with pMS and relapses. 24 However, its widespread use is limited and this procedure is not approved in some countries. 23
Intravenous immunoglobulin
Evidence of the effect of intravenous immunoglobulin (IVIg) on relapses in aggressive MS is very limited. A small, randomized, double-blind, placebo-controlled pilot study in 19 MS patients compared the efficacy of IVIg given with IV corticosteroids to IV corticosteroids alone to promote recovery from moderate to severe relapses. The primary outcome was the EDSS level at 4 weeks. No statistically significant differences were found in the median change in EDSS (p = 0.81). Furthermore, the median time to improvement of at least one point on the EDSS was 14 days in both groups (p = 0.95). 25
Based on these published results, limited evidence appears to support the use of PLEX and IA for corticosteroid-refractory MS relapses. However, IVIg appears to have no effect in severe relapses (Table 1).
Disease-modifying treatments
Tables 2–4 summarize the different studies assessing treatment in patients with aggressive MS. Definitions used in the cited studies for aggressive rMS vary from exclusively clinical or radiological to combinations of both. Many of these studies compare disease-modifying treatments (DMTs) to placebo or to first-line treatments.26–35 In the following text, we focus on studies with drug–drug comparisons; studies assessing naïve patients who started a high-efficacy DMT and studies assessing patients who switched from first-line to high-efficacy DMT or from one high-efficacy DMT to another. Of note, there are no specific studies on treatment for aggressive pMS.
Importantly, although not available at the time of the workshop, data since have shown a superiority of monoclonal antibodies (Mabs) over oral DMTs. Most of said studies were not included in this review because: (1) they do not assess patients with aggressive MS, (2) it is impossible to determine if the included subjects have aggressive MS according to the baseline characteristics or (3) they do include a subgroup with aggressive MS but it is not evaluated individually.
Oral and intravenous DMTs
Dimethyl fumarate (DMF) was assessed in a subgroup analysis of patients with high clinical or radiological activity in the DEFINE and CONFIRM trials.26,27 However, results were not compared to other DMTs in this analysis.
Fingolimod has been compared to other DMTs and showed a greater effect on the annualized relapse rate (ARR) compared with interferon (IFN)-β1a in patients with high clinical or radiological activity. 30 However, fingolimod might not be as effective as Mabs and its superiority over DMF is not clear. A propensity score-matching analysis assessed no evidence of disease activity (NEDA-3) at 2 years in two patient datasets after starting natalizumab, fingolimod or self-injectables: one study included patients with high clinical activity despite first-line DMTs and one included highly active, treatment-naïve patients. 36 When comparing natalizumab and fingolimod, the proportion of patients with NEDA-3 was greater for natalizumab than fingolimod in the first dataset, whereas in the second, the difference did not reach statistical significance, probably due in part to the smaller size of that cohort. 36
A systematic review and meta-analysis of highly active rMS and rapidly evolving severe MS explored the efficacy of fingolimod relative to DMF and natalizumab. 37 In the highly active relapsing–remitting multiple sclerosis (RRMS) population, a trend favoured fingolimod over DMF in reducing the ARR and confirmed disability progression. In the rapidly evolving severe MS population, patients on fingolimod had a non-statistically significant increase in the ARR and in confirmed disability progression compared to patients on natalizumab. 37 This meta-analysis has limitations due to its retrospective assessment and was not powered to indicate differences only in subjects with a highly active or severe disease.
Conversely, a study based on a national registry using propensity score matching in patients switching to second-line DMTs after breakthrough disease while on IFN-β or glatiramer acetate did demonstrate natalizumab was more effective than fingolimod in reducing the number of relapses and improving disability. 38 Although the selection criteria for this study did not restrict patient selection to cases with highly active disease, the mean (SD) number of relapses in the year prior to inclusion was 2.8 (2.0) for patients switching to natalizumab and 2.7 (3.1) for patients switching to fingolimod, which might be considered a relatively high relapse rate for included subjects. Another hospital-based study also demonstrated natalizumab was superior to fingolimod in inducing regression of disability and NEDA at 2 years. 39 There were, however, no significant differences between medications when comparing the clinical components of NEDA individually. These results could be due to the small sample size after propensity score matching and dropouts in the fingolimod group due to breakthrough disease.
Post hoc subgroup analyses of the CLARITY clinical trial in patients with high disease activity compared cladribine to placebo.31,32 A recent analysis suggested that patients with highly active MS have a better response to cladribine than those with a milder disease course. This analysis classified patients into two overlapping, categorical subgroups: the high relapse activity (HRA) and the HRA plus disease activity on treatment (HRA + DAT) subgroups. 32 During cladribine treatment, the ARR was lower in HRA versus non-HRA patients and in HRA + DAT versus non-HRA + DAT patients, although without reaching statistical significance. Similarly, the risk of new Gd+ lesions decreased slightly in patients with a more active disease. 32
A prospective hospital-based study assessed 70 patients with highly active MS treated with natalizumab, of whom 97.1% had received a previous DMT. The proportion of patients free of clinical and overall disease activity was 60.0%–70.0% at 1 year but decreased to approximately 50.0% after 2 years. Conversely, freedom from radiological activity was consistently high throughout the study period. 40
Alemtuzumab has been compared to IFN-β1a in a post hoc and subset analysis of the CAMMS223 trial. Results favoured alemtuzumab in patients with frequent relapses. 41 In a single small study of alemtuzumab as rescue therapy after mitoxantrone in patients with rMS and SPMS, some subjects showed EDSS improvement and NEDA. 42
Finally, ocrelizumab has shown increased treatment benefits relative to IFN-β1a by reducing disease activity and disability progression in a subgroup of rMS patients with highly active disease. 43 Similarly, rituximab has proven effective in patients with rMS with persistent disease activity despite treatment with other high-efficacy DMTs, such as fingolimod, natalizumab or mitoxantrone 44 (Tables 2 and 3).
Autologous haematopoietic stem cell transplantation
Autologous haematopoietic stem cell transplantation (aHSCT) is used to induce long-term immunosuppression and immune reconstitution in aggressive forms of MS. To date in MS, it has been mostly used as an escalation therapy approach when more than one first- or second-line DMTs has failed.
Early evidence on the efficacy of aHSCT came as small case series showing a significant improvement in EDSS and reduction in number of relapses. 45 Results from two Phase 2 clinical trials (one including both rMS and pMS patients and one with rMS patients only) support the efficacy of aHSCT in patients with high clinical activity. In these studies, two-thirds of subjects were free of disease activity at 3 and 5 years and no treatment-related mortality was reported in one of the trials.46,47 Another Phase 2 trial compared aHSCT and mitoxantrone in rMS and pMS patients with high clinical and radiological activity, using cumulative number of new T2 lesions after 4 years as primary endpoint. 48 Compared to mitoxantrone, aHSCT reduced the number of new T2 lesions by 79.0% and also had an effect on Gd+ lesions and ARR, but no difference was found in disability progression. 48 A long-term, multicentre, observational, retrospective cohort of rMS and pMS patients who received aHSCT showed that the overall 5-year probability of progression-free survival was 46.0%, and the size effect was greater in RRMS than in SPMS patients. Importantly, factors increasing the risk of overall worse survival after aHSCT are older age, pMS, more than two previous DMTs and a high EDSS. 49
These findings are supported by a meta-analysis evaluating the rate of disease progression and NEDA after aHSCT. At 5 years, the pooled rate of progression was 23.3% and NEDA was observed in 67.0% of patients. In general, progression-free survival in patients has increased in studies post-2005, in younger patients with EDSS ⩽ 5.5 and in rMS. 50 Finally, when aHSCT is compared to first- and second-line DMTs tested in Phase 3 trials, NEDA at 2 years was greater in patients undergoing aHSCT than in those receiving placebo or DMTs. However, these results should be interpreted with caution due to the absence of direct comparisons. 51 aHSCT is still considered experimental in MS, with a need to demonstrate a sound risk/benefit profile in a well-defined target population (Table 4).
Mitoxantrone
The efficacy of mitoxantrone has been assessed against methylprednisolone or placebo in studies, including both RRMS and SPMS.52,53 One of the rare trials including a well-defined highly active population studied the effect of short-term immunosuppression with mitoxantrone in patients with aggressive RRMS, defined as ⩾ 2 relapses or an EDSS increase ⩾ 2 points in the 12 preceding months, ⩾ 1 Gd-enhancing lesion and baseline EDSS between 2.5 and 5.0. The time to worsen by ⩾ 1 point in EDSS at 3 months was delayed in patients who received mitoxantrone plus methylprednisolone for 6 months before switching to IFN-β, compared to patients receiving IFN-β plus methylprednisolone for 6 months who then continued treatment with IFN-β alone. 54 Nevertheless, use of mitoxantrone has declined due to serious adverse events and it should be considered only in individual cases.
Special situations and populations
Pregnancy
Pregnancy risks should be discussed with patients and spouses before starting any DMT. 55 Information on treatment of patients with highly active MS during pregnancy is limited. Most data come from studies and observations on individuals with a broad spectrum of MS phenotypes and severities. Therapies which can be administered throughout pregnancy or those which are stopped at conception but have lasting effects throughout pregnancy should be considered.
Immune reconstitution treatments administered in short courses, such as alemtuzumab, cladribine and probably rituximab, offer a window of opportunity in patients with aggressive MS planning a pregnancy (typically 4–6 months after the last dose, with some authors suggesting 1 month in the case of rituximab).56–59 To minimize the risk of recurrent disease activity in patients stable while using natalizumab, continuing treatment until the beginning of the third trimester or throughout pregnancy should be considered, as the haematological changes reported in exposed newborns are temporary and not clinically relevant.55,60 In the rare occurrence of aggressive MS developing in a pregnant woman, natalizumab or anti-CD20 therapies might be considered, but evidence of their safety and efficacy in such a situation is scant. This is an area for future research. Fingolimod is contraindicated not only due to the risk of recurrent disease activity after stopping treatment, but mainly due to the increased risk of major birth defects in exposed infants. 61
After delivery, early reintroduction of DMTs is advisable if prior therapies do not have long-lasting therapeutic effects.62,63 In case of relapses, PLEX has been used successfully during pregnancy, 64 but caution should be used since it can aggravate IRIS-like phenomena following natalizumab discontinuation.65,66 There is no specific evidence supporting the use of IA or IVIg as alternatives to PLEX in pregnant women with highly active MS.
Children
Severe relapses in children with MS have been treated with repeated corticosteroid boluses, PLEX, IVIg, rituximab and cyclophosphamide with varying degrees of benefit.67–69 However, inclusion of patients with demyelinating diseases other than MS cannot be ruled out in some of these studies, so their relevance to a paediatric MS population may be limited.
A retrospective single-centre study identified paediatric patients with highly active disease according to the definition in German guidelines 70 (⩾ 1 attack within the previous year and ⩾ 9 T2 lesions or ⩾ 1 Gd+ lesion while under first-line DMT. At diagnosis, ⩾ 2 attacks and EDSS worsening within the last 12 months and ⩾ 1 Gd+ lesion or a significant increase in T2 lesions within the last 6–12 months). The 12-month period before and after treatment initiation with natalizumab or fingolimod was studied. After 1 year of treatment, relapse rate was reduced by 95.2% in natalizumab-treated patients and 75.0% in fingolimod-treated patients. Significant reductions were also observed in MRI parameters of disease activity. When compared to a historical cohort of patients with mild-to-moderate disease activity treated with first-line therapies, patients on natalizumab or fingolimod had a 44.0% reduction in mean EDSS. 71
Other than these, there are no treatment studies specifically addressing highly active disease in children. Nevertheless, most children present with a higher disease activity than adults and can be considered as having aggressive MS simply by their natural history. In the PARADIGMS clinical trial 72 in paediatric MS, fingolimod was associated with a lower ARR (relative difference of 82.0%) and with less accumulation of MRI-detected brain lesions than IM IFN-β1a. Evidence relating to the use of rituximab in children is minimal and not focused in highly active MS. 73
In summary, children with aggressive MS could benefit from initiating a more efficacious DMT from the start.69,74 Such a strategy takes the generally higher disease activity in this population into account, but it will have to be prospectively studied. The long-term impact of such therapies in children, including clinical and immunological developmental milestones, remains unknown.
Senior patients
There are no data regarding DMT in senior patients (> 65 years of age at time of treatment) with aggressive MS. In this age group, the safety and tolerability of highly effective DMTs in very active cases may be affected by MS- and age-related comorbidities.10,75–77 Given that senior patients are usually excluded from clinical trials, encouraging their representation in prospective trials would increase the available evidence but, to date, real-world data (registries, observational cohorts) may be the best source of information for this patient group.
How to manage and prevent complications of high-efficacy treatments
Risk-minimizing strategies before, during and after treatment with high-efficacy DMTs are reviewed in detail elsewhere. 78 It is important to note that the time frame to implement such strategies is likely narrower in patients with aggressive MS. In addition, the occurrence and management of adverse events may be different for aggressive and non-aggressive MS depending on the comorbidities that may be associated with more severe MS and the adverse event profile of individual treatments. A list of some adverse events related to the immunosuppressive effects of these DMTs is shown in Supplemental Table 2.
Beyond treatment-related AEs, all patients should receive vaccinations as indicated in their respective regions, with special considerations depending on the type of vaccination and the MS treatment plan. A 4 to 6-week interval is typically recommended between administration of live-attenuated vaccines and initiation of high-efficacy treatments.79,80 Until new data emerge supporting the safety of live-attenuated vaccines in patients treated with immunosuppressive therapies, such vaccines are contraindicated during therapy and for a variable period after treatment termination, usually ranging from 2 to 6 months. 81 Data support the effectiveness of vaccinations (sometimes reduced compared to placebo) during treatment with natalizumab, fingolimod or DMF, indicating that inactivated, subunit and toxoid vaccines can be administered in such cases. 81
Achievements, gaps in knowledge and future perspectives
Considering available evidence to date regarding treatment in patients with aggressive MS, direct data are limited. The PLEX randomized, sham-controlled trial for severe attacks 15 and the mitoxantrone and methylprednisolone randomized multicentre study in rMS 54 were the only trials we identified that included patients with an aggressive disease. However, the former was not limited to patients with MS and the latter included patients with different MS phenotypes.
Therefore, many open issues remain. An important limitation to address the questions on treatment is the lack of a unified definition of aggressive MS. Before any consensus on treatment of aggressive MS can be obtained, it will be particularly important to develop a unified definition of aggressive disease. To the extent possible, paraclinical and clinical biomarkers may aid stratify the more severe cases of both relapsing and pMS phenotypes. As mentioned before, two studies have recently proposed EDSS ⩾ 6.0 at 10 years as a definition of aggressive MS,8,9 but this proposal needs to be validated in other cohorts or registries. In addition, it is important to assess whether other biomarkers of aggressive disease (i.e. potentially neurofilament light chain levels, atrophy measures) may have an added value to NEDA-3 and other scores of breakthrough disease.
Open issues also remain regarding treatment of severe attacks: does oral methylprednisolone act as quickly and effectively as the IV form when an equivalent dose is used? Is 1 g/day the best dose in these cases? When should treatment be escalated to PLEX? Can natalizumab or anti-CD20 Mabs be used as treatments of severe attacks?
Concerning the use of DMTs, an escalation paradigm does not appear to be a good option in patients with aggressive MS. Based on current data, their window of therapeutic opportunity is narrow, as response to DMTs is better early in the disease course and in younger patients with minimal disability.6,82 Rather than escalating, starting a high-efficacy treatment in these instances would likely be a better option to limit the possibilities of breakthrough disease. Nevertheless, the evidence for conducting such approach is limited because pivotal trials were not focused in patients with aggressive MS. As for the post hoc analyses from the DMTs pivotal clinical trials, they are limited to comparisons with placebo or self-injectables, short term and underpowered to demonstrate any significant differences among DMTs in the more aggressive subpopulations. In the case of aHSCT, the most recent studies show that highly active patients with rMS are good candidates for this treatment, but such studies lack active comparators.
Consequently, more rigorous prospective studies are needed to confirm which treatments are the best option for patients with aggressive MS. Examples include the ongoing trials comparing aHSCT to high-efficacy treatments in patients with breakthrough disease despite first- or second-line DMTs. 83 When considering a trial design for aggressive MS, a first step would be to consider the features that best define this phenotype early in the disease course to inform the inclusion criteria. A second step would consist in identifying a medium-term (2–3 years) endpoint related to, for example, EDSS 6.0 at 10 years, sensitive enough to occur frequently in the control group. The next steps would be selecting the active comparator the control group will receive and hypothesizing the expected reduction in outcome events with the more efficacious DMT. It is then that the sample size can be calculated. Finally, a decision on performing a conventional clinical trial or a pragmatic trial can be made according to the sample size.
It is important to consider that head-to-head comparative studies require very large sample sizes to demonstrate the differential efficacy and safety using the current outcome measures. Conducting pragmatic trials is a different approach that evaluates the effectiveness of an intervention in the real-world setting. 84 These trials are designed to test interventions in the full spectrum of everyday clinical settings to maximize applicability and generalizability. Pragmatic trials measure mostly patient-centred outcomes, whereas conventional trials focus on measurable symptoms or biomarkers. 85 Examples of such trials are TREAT-MS and DELIVER-MS, which compare two treatment paradigms: early highly effective therapy versus escalation of therapy. 86 However, these two trials are not designed specifically for aggressive MS.
In addition, evidence from long-term follow-up of patients included in clinical trials and large, real-world cohorts assessing different subgroups of patients with MS, including pMS and special populations with aggressive MS, may provide useful data when controlled trials are not feasible.
Finally, we are aware that limitations on treatment access to highly efficacious DMTs vary widely from country to country, mostly due to financial restrictions. We hope papers such as this one may help MS specialists provide information about the usefulness of starting a highly efficacious DMT in patients with aggressive MS to their local regulatory agencies.
Supplemental Material
MSJ924595_supplemental_table_1 – Supplemental material for Aggressive multiple sclerosis (2): Treatment
Supplemental material, MSJ924595_supplemental_table_1 for Aggressive multiple sclerosis (2): Treatment by Georgina Arrambide, Ellen Iacobaeus, Maria Pia Amato, Tobias Derfuss, Sandra Vukusic, Bernhard Hemmer, Lou Brundin and Mar Tintore in Multiple Sclerosis Journal
Supplemental Material
MSJ924595_supplemental_table_2 – Supplemental material for Aggressive multiple sclerosis (2): Treatment
Supplemental material, MSJ924595_supplemental_table_2 for Aggressive multiple sclerosis (2): Treatment by Georgina Arrambide, Ellen Iacobaeus, Maria Pia Amato, Tobias Derfuss, Sandra Vukusic, Bernhard Hemmer, Lou Brundin and Mar Tintore in Multiple Sclerosis Journal
Footnotes
Acknowledgements
The authors thank ECTRIMS for financial and administrative support of the focused workshop that served as the basis for this paper and all workshop attendees (![]() ) for their participation. All attendees had the opportunity to review and comment upon an advanced draft of the manuscript prior to submission. They also thank Dr Stephen C. Reingold for assistance with manuscript writing and editing, supported by ECTRIMS.
) for their participation. All attendees had the opportunity to review and comment upon an advanced draft of the manuscript prior to submission. They also thank Dr Stephen C. Reingold for assistance with manuscript writing and editing, supported by ECTRIMS.
Declaration of Conflicting Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: G.A. has received compensation for consulting services or participation in advisory boards from Sanofi, Merck and Roche; research support from Novartis; travel expenses for scientific meetings from Novartis, Roche and Stendhal; and speaking honoraria from Sanofi and Merck. E.I. has honoraria for advisory boards or lecturing for Sanofi Genzyme and Merck outside the submitted work. M.P.A. has received research grants and personal fees as a speaker and member of advisory boards by Biogen, Sanofi Genzyme, Merck Serono, Novartis, Roche and Teva outside the submitted work. T.D. reports grants from Novartis and Biogen. His institution received financial support from Novartis, Biogen, Roche, Merck Serono, Celgene, Sanofi Genzyme, MedDay, GeNeuro, Mitsubishi Pharma and Actelion for his activities as steering committee or advisory board member or consultant. S.V. has received grants, personal fees and non-financial support from Biogen, Celgene, Geneuro, Genzyme, Medday, Merck Serono, Novartis, Roche, Sanofi and Teva outside the submitted work. B.H. has served on scientific advisory boards for Novartis and as DMSC member for AllergyCare and TG therapeutics, he or his institution have received speaker honoraria from Desitin, holds part of two patents: one for the detection of antibodies against KIR4.1 in a subpopulation of MS patients and one for genetic determinants of neutralizing antibodies to interferon. L.B. has received honoraria for advisory boards for Biogen, Sanofi Genzyme, Novartis, and Teva and has received lecturing fees from Biogen, Novartis, Teva and Sanofi Genzyme outside the submitted work. M.T. has received compensation for consulting services and speaking honoraria from Almirall, Bayer Schering Pharma, Biogen Idec, Genzyme, Merck Serono, Novartis, Roche, Sanofi Aventis and Teva Pharmaceuticals. M.T. is the co-editor of Multiple Sclerosis Journal–ETC.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The workshop on which the manuscript is based was supported in its entirety by the European Committee on Treatment and Research in Multiple Sclerosis (ECTRIMS).
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.