
Abstract
Background/objective:
Observational clinical data from cladribine-treated patients with relapsing forms of multiple sclerosis (MS) were recorded in the Australian MS registry powered by the MSBase registry platform (5-year follow-up) and analysed to complement information from the pivotal cladribine clinical trials in MS.
Methods:
A cohort of 90 cladribine-treated patients with follow-up data reported by treating physicians and recorded in the Australian MSBase registry (database lock February 2016) were examined. Clinical data included Expanded Disability Status Scale (EDSS) scores, relapses and other disease-modifying drugs (DMDs) administered before and after cladribine treatment.
Results:
Mean age on starting cladribine was 47 years; mean age at MS onset was 34 years, and median baseline EDSS score was 5.25. Disability trajectories in patients with sufficient follow-up suggested an overall increasing trend prior to cladribine treatment which was reduced during the 2-year post-treatment. Approximately 80% of patients were EDSS progression-free, 65% remained relapse-free after 2 years and median time to next DMD was 1.7 years.
Conclusion:
These observational data suggest a disease-modifying effect in this cohort of relapsing MS patients characterised by older and more disabled patients. Since these data represent a single-arm cohort, clinical trials and larger comparative post-marketing studies are needed to validate and extend these findings.
Keywords
Introduction
Cladribine tablets 10 mg (MAVENCLAD®; cumulative dose 3.5 mg/kg over 2 years) are approved in several countries, including Australia, for the treatment of selected adult patients with relapsing–remitting multiple sclerosis (RRMS). 1 Each treatment course consists of 2 treatment weeks, one at the beginning of the first month and one at the beginning of the second month of the respective treatment year. The treatment schedule in each week consists of 4 or 5 days on which a patient receives 10 or 20 mg (one or two tablets) depending on body weight, as a single daily dose.
In the CLARITY study, cladribine tablets demonstrated significant reductions versus placebo in relapse rates, risk of disability progression and magnetic resonance imaging (MRI) measures of disease activity in patients with RRMS.2,3 In the CLARITY Extension study, the durable clinical efficacy of cladribine was demonstrated when given at a cumulative dose of 3.5 mg/kg over 2 years, followed by placebo treatment for a further 2 years. 4 Treatment with cladribine tablets also demonstrated a reduced risk of conversion to clinically definite multiple sclerosis (MS) versus placebo in patients with a first clinical demyelinating event, and a reduced risk of next attack or 3-month confirmed disability progression, in patients with early MS.5,6 Efficacy has been demonstrated across subgroups of patients with RRMS, including those with high disease activity and in patients with active disease, despite prior treatment with interferon β.7–9
Cladribine tablets were first registered in Australia on 2 September 2010 and made available by a Patient Familiarisation Program (PFP) that is provided by pharmaceutical companies launching newly licenced medicines while Pharmaceutical Benefits Scheme (national payer) listing is pending. 10 Following a regulatory decision in Europe, the product was subsequently withdrawn from use in Australia in late 2011. During the period when cladribine was commercially available in Australia, 144 patients with relapsing MS (RMS) received the product as part of the Australian PFP. 11
Data from 90 of these patients were captured by clinics participating in the Australian MS registry, as part of the international observational MSBase registry. MSBase records a minimum dataset of long-term follow-up, including disease-modifying drug (DMD) use and disease outcomes (relapses, Expanded Disability Status Scale (EDSS) score). 12 Enrolment and follow-up in the MSBase registry is not dependent on drug exposure. The cohort of patients included in the MSBase registry who received treatment with cladribine formed the basis for a previously published propensity score–matched analysis to compare cladribine with fingolimod, natalizumab, and interferon β. 11 This comparison suggested cladribine efficacy on relapse is similar to fingolimod, and on disability accrual is similar to interferon β and fingolimod. A potentially superior recovery from disability may be associated with cladribine, relative to interferon, fingolimod, and natalizumab. 11
Most of the existing knowledge about the use of cladribine tablets in MS comes from phase 3 clinical trials. With cladribine gaining approvals in countries around the world, observational data can provide outcome information on routine use. Here, we report the clinical outcomes from patients treated with cladribine with data recorded in the Australian MSBase registry, in particular, those who received treatment under the PFP.
Materials and methods
Ethics statement
MSBase is registered with the World Health Organization International Clinical Trials Registry Platform (ID ACTRN12605000455662). 12 This study was approved by the Melbourne Health Human Research Ethics Committee and either approved by participating sites’ institutional review boards or granted exemptions according to local regulations. All patients provided written informed consent.
Patients and data collection
This study included the cohort of patients in Australia with a diagnosis of RMS who were treated with cladribine and with data from follow-up recorded in the MSBase registry database. The database lock occurred in February 2016.
Clinical data are presented as reported by the patients’ treating physician and recorded in the MSBase registry database. Clinical data examined included EDSS scores, relapses, and other DMDs administered before and after treatment with cladribine. To qualify for the analysis of disability outcomes, patients needed to have at least two EDSS assessments, including a baseline score between 1 year prior and 3 months after first administration of cladribine.
Diagnosis of RMS at the time of enrolment in the PFP was determined by the treating neurologist. 13 In a small number of patients (18), a diagnosis of secondary progressive multiple sclerosis (SPMS) was assigned to the patients retrospectively, in keeping with the delay in definitive identification of an SPMS phenotype.14,15 Adverse event data are not systematically captured in the MSBase registry and are therefore not reported here.
Analyses
All statistical analyses are descriptive. Inferential or comparative statistics were not conducted. Disability progression was defined as increase in EDSS by the following: (a) 1.5 steps if baseline EDSS = 0; (b) 1 step if baseline EDSS was between >0 and <6; or (c) 0.5 step if baseline EDSS ⩾ 6, confirmed over at least 6 months in the absence of a relapse and sustained for the remainder of follow-up. The probability of a relapse or confirmed disability progression, end of follow-up, and switch to another DMD were quantified with a Kaplan–Meier estimator. The mean disability trajectory was interpolated using serial EDSS scores with a fitted spline, applying a local kernel for smoothing, with a t distribution used to estimate the error margin.
Results
Patient population
There were 144 patients in the PFP treated between January and 12 July 2011. In total, 90 patients with MS from 13 sites across Australia who had been treated with cladribine were identified in the MSBase registry, of whom 87 were treated as part of the PFP. Due to the withdrawal of the commercially available product before the second year of treatment was due, the 87 patients in the PFP only received the first year of cladribine treatment. Three patients were treated with cladribine outside the PFP between 1995 and 2010 (one received off-label intravenous cladribine on compassionate grounds); the dose of cladribine received by these three patients is unknown.
Of the 86 patients with at least one post-cladribine EDSS, 20 did not have baseline EDSS recorded. Consequently, there were 66 patients with at least two EDSS assessments in the analysis period, including a baseline score, for whom outcomes are presented (Figure 1).
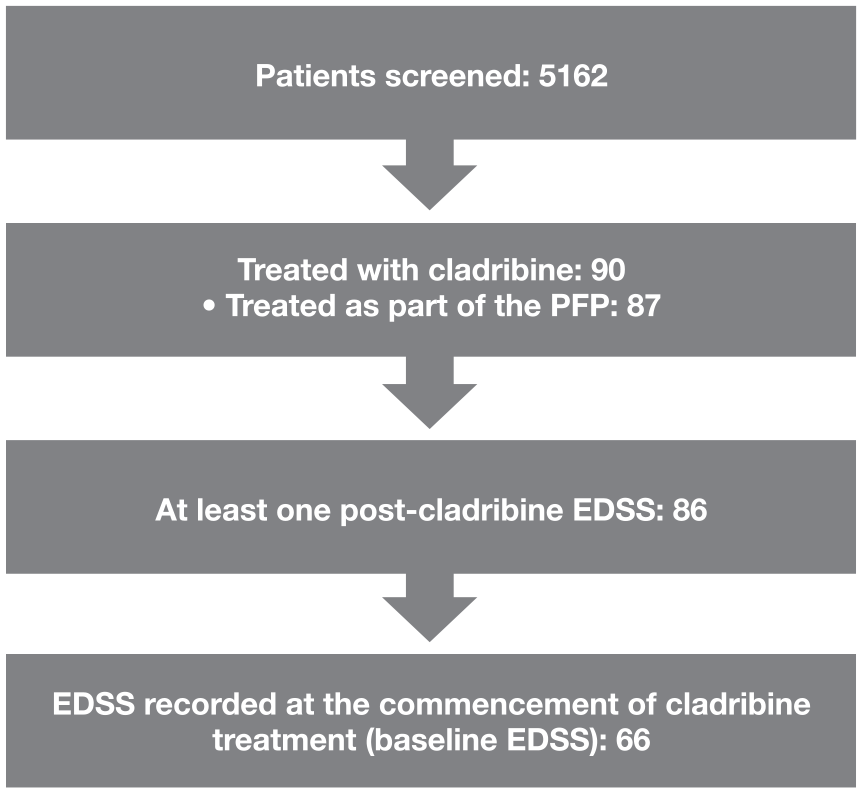
Patient disposition
Baseline demographics and clinical characteristics of the overall patient population (N = 90) and those with sufficient EDSS follow-up (N = 66) are shown in Table 1. Overall, mean age at onset of MS was 34 years (standard deviation (SD) ± 11), and mean duration of MS was 13 years (SD ± 9). At the time of first cladribine exposure, 70 patients had RRMS, and 18 had SPMS (Table 2). Disease classifications could have been updated by the clinicians since the time of the PFP in 2011.
Demographics and baseline characteristics of patients receiving cladribine in the Australian substudy of the MSBase registry cohort and 66 patients with sufficient EDSS follow-up*.
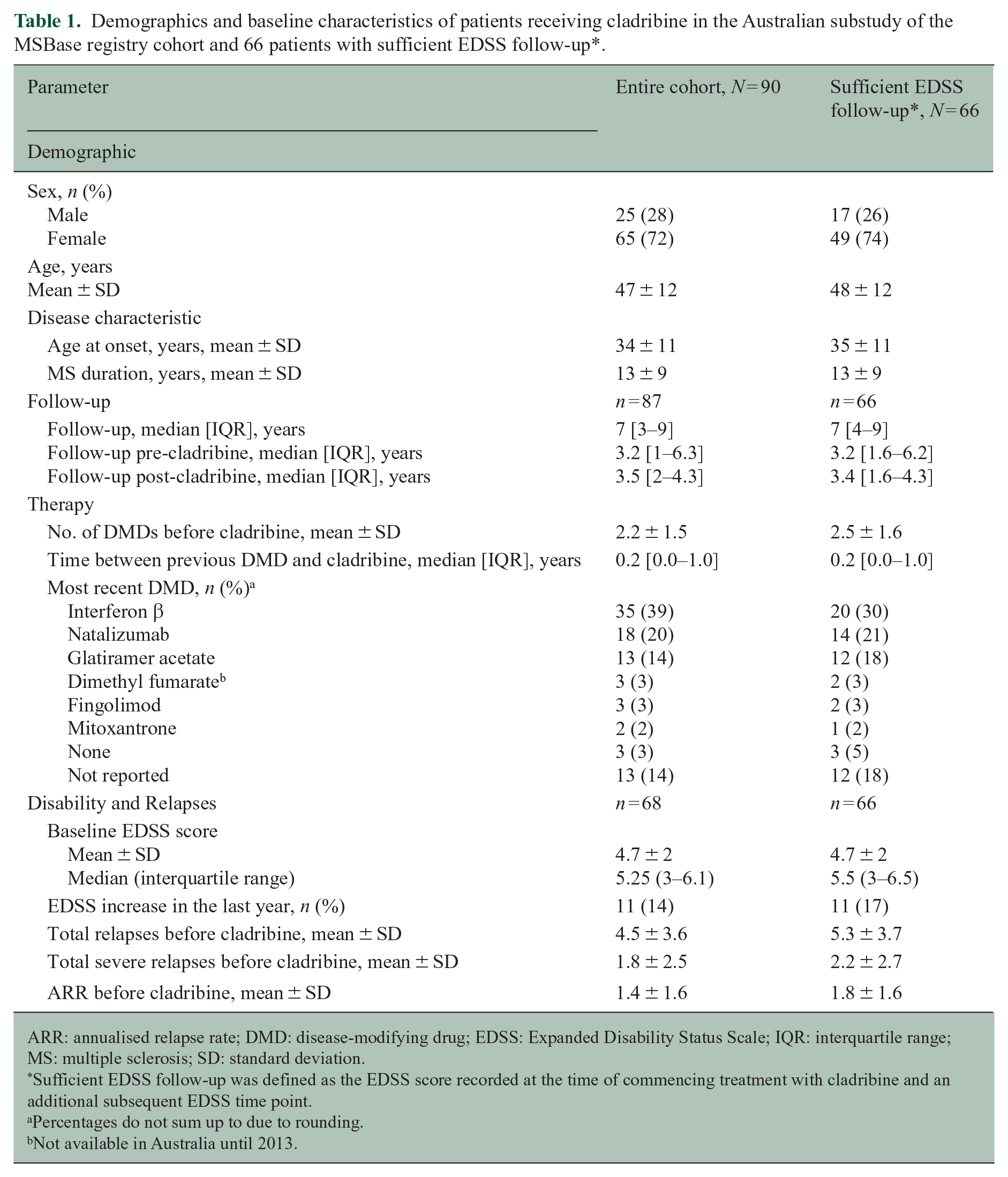
ARR: annualised relapse rate; DMD: disease-modifying drug; EDSS: Expanded Disability Status Scale; IQR: interquartile range; MS: multiple sclerosis; SD: standard deviation.
Sufficient EDSS follow-up was defined as the EDSS score recorded at the time of commencing treatment with cladribine and an additional subsequent EDSS time point.
Percentages do not sum up to due to rounding.
Not available in Australia until 2013.
Subgroups of patients with RRMS or SPMS at the time of first cladribine exposure.

ARR: annualised relapse rate; RRMS: relapsing–remitting multiple sclerosis; SPMS: secondary progressive multiple sclerosis; SD: standard deviation.
Sufficient EDSS follow-up was defined as the EDSS score recorded at the time of commencing treatment with cladribine and an additional subsequent EDSS time point.
Patients in the overall cohort had a median baseline EDSS of 5.25 (interquartile range (IQR) 3.0–6.1). For the RRMS cohort (those not retrospectively diagnosed with progressive MS; n = 70), the median EDSS was 4.5 (IQR 2.5–6.0). DMD use prior to treatment with cladribine in the overall cohort was reported in 74 (82%) patients, with a mean of 2.2 prior DMDs (SD ± 1.5). The most common recent prior treatments were interferon β, natalizumab, and glatiramer acetate.
Patient follow-up
Overall, patients were followed up for a median (IQR) of 3.5 (2–4.3) years after receiving cladribine (Table 1). Patients (n = 68; data missing n = 22) had a median (IQR) of five follow-up visits (1–7.5), with a median (IQR) visit frequency of 1.5 per year (1–2.4). Approximately 80% of patients with data from two EDSS assessments were still followed at 2 years after receiving cladribine (Figure 2).
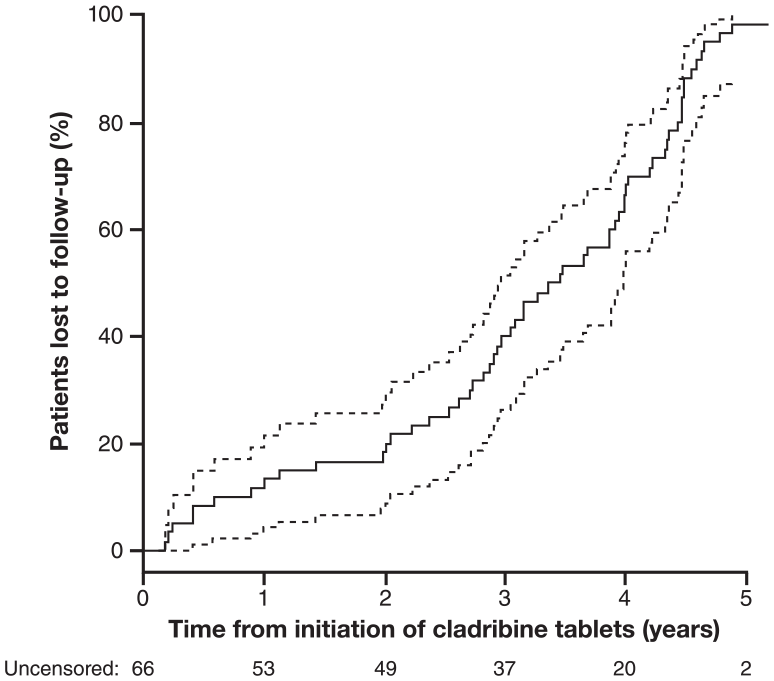
Patient follow-up after cladribine treatment in patients with sufficient EDSS follow-up* (n = 66 at Time 0).
Relapses
Approximately 65% of patients in the overall cohort (n = 66 with sufficient follow-up data) were relapse free 2 years after treatment with cladribine (Figure 3). Relapse frequency remained stable during the 2 years prior to, and 2 years after, commencement of cladribine (baseline data not shown). Almost identical observations were made for the RRMS cohort. In the cohort with RRMS and sufficient follow-up data (n = 51), the mean annualised relapse rate (ARR) prior to cladribine treatment was 1.8 (SD ± 1.7) and 0.31 (SD ± 0.52) after cladribine treatment. In the cohort with SPMS and sufficient follow-up data (n = 14), mean ARR was 1.6 (SD ± 1.2) before cladribine treatment and 0.30 (SD ± 0.52) after cladribine treatment. For both cohorts, ARR was calculated over the entire available follow-up post-commencement of cladribine treatment and therefore influenced by short follow-up.
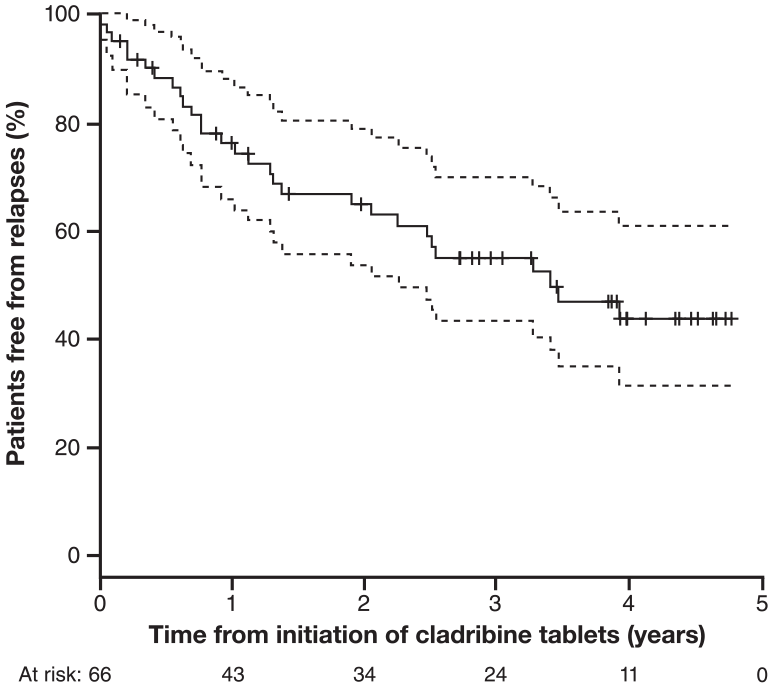
Freedom from relapse after cladribine treatment in patients with sufficient EDSS follow-up* (n = 66 at Time 0).
Disability (EDSS) progression
Approximately 80% of patients with adequate post-cladribine follow-up (n = 66) were free from EDSS progression 2 years after treatment with cladribine (Figure 4(a)). EDSS trajectories for the study population with RRMS prior to and after treatment with cladribine are shown in Figure 4(b). In the subpopulation retrospectively categorised with SPMS, the EDSS trajectory continued to increase after the treatment with cladribine (Figure 4(c)). Of the 14 patients with SPMS and sufficient EDSS information during follow-up, EDSS scores decreased in 1, increased in 1 and remained unchanged in 12 patients during the initial year post-cladribine. Annualised change in the EDSS-time curve reached a median of +0.8 EDSS years (IQR +0.3 to +1.1) in the SPMS cohort. The differences between the EDSS trajectories 2 years before and after cladribine treatment appeared to be relatively less pronounced in patients with SPMS than in patients with RRMS.
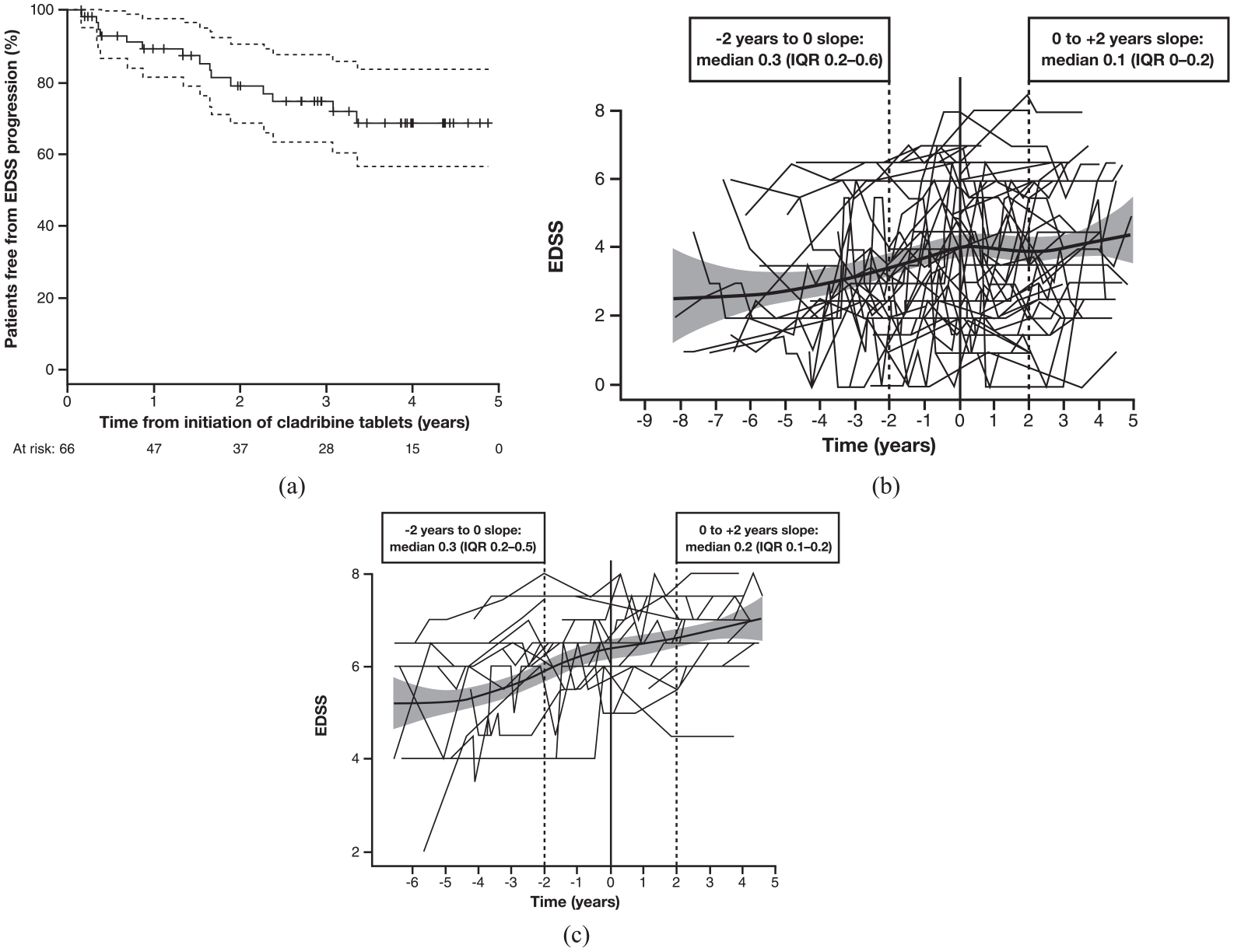
(a) Freedom from EDSS progression after cladribine treatment, and EDSS trajectories for patients with either (b) RRMS or (c) SPMS in patients with sufficient EDSS follow-up* (n = 66 at Time 0).
Subsequent DMD after treatment with cladribine
In the overall cohort, 62 of 90 patients (69%) received another DMD following cladribine treatment during the reported follow-up period (Table 3). Overall, 45 (73%) patients who switched did so before a relapse and 17 (27%) experienced relapses prior to switching to another DMD. The time of switching to other DMDs is shown in Figure 5. The median (95% confidence interval (CI)) time to next DMD was 1.7 years (1.36–2.28).
Patient exposure to other DMDs after cladribine in the overall cohort and subgroups of patients with RRMS or SPMS at the time of first cladribine exposure.
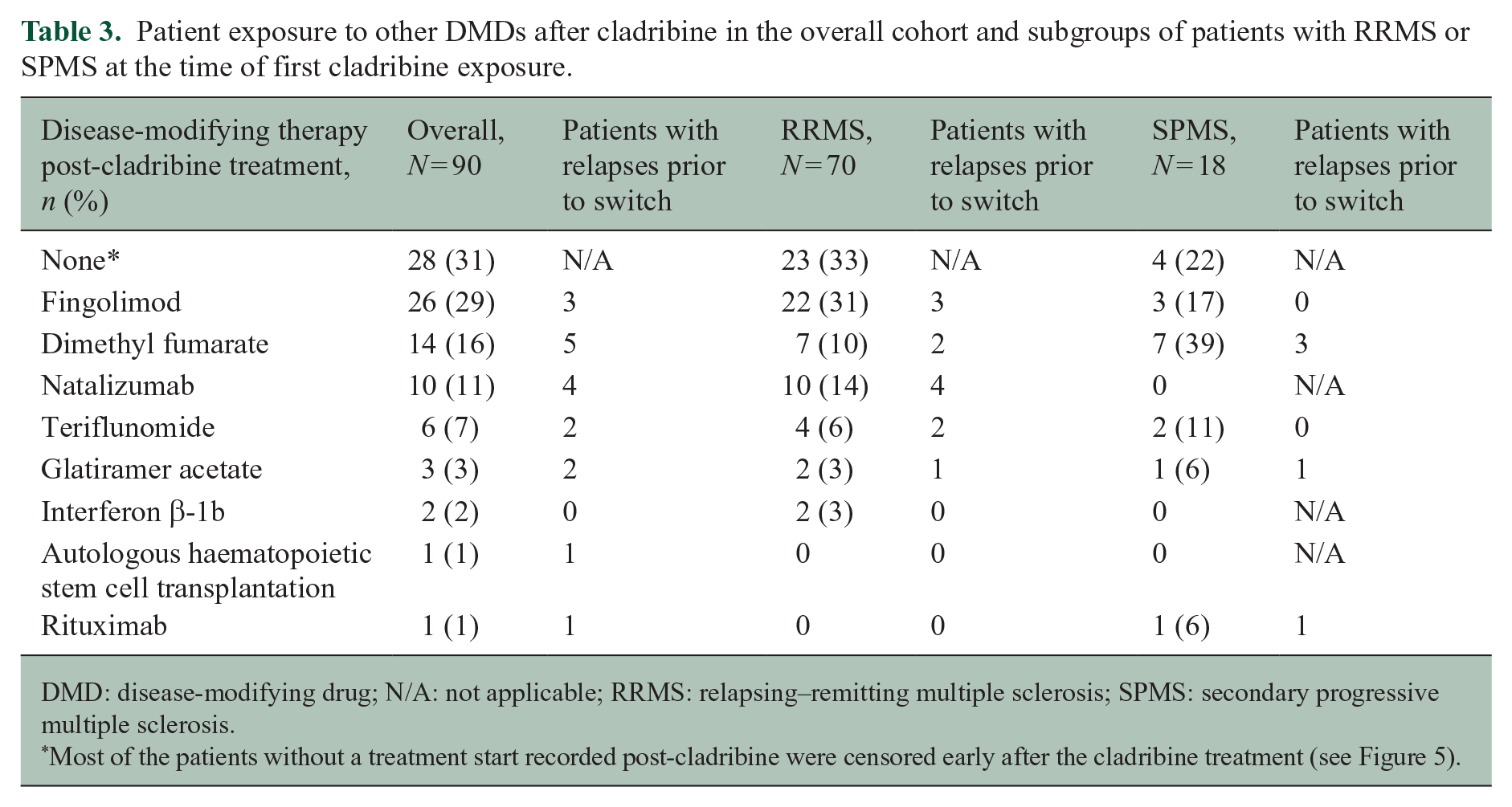
DMD: disease-modifying drug; N/A: not applicable; RRMS: relapsing–remitting multiple sclerosis; SPMS: secondary progressive multiple sclerosis.
Most of the patients without a treatment start recorded post-cladribine were censored early after the cladribine treatment (see Figure 5).
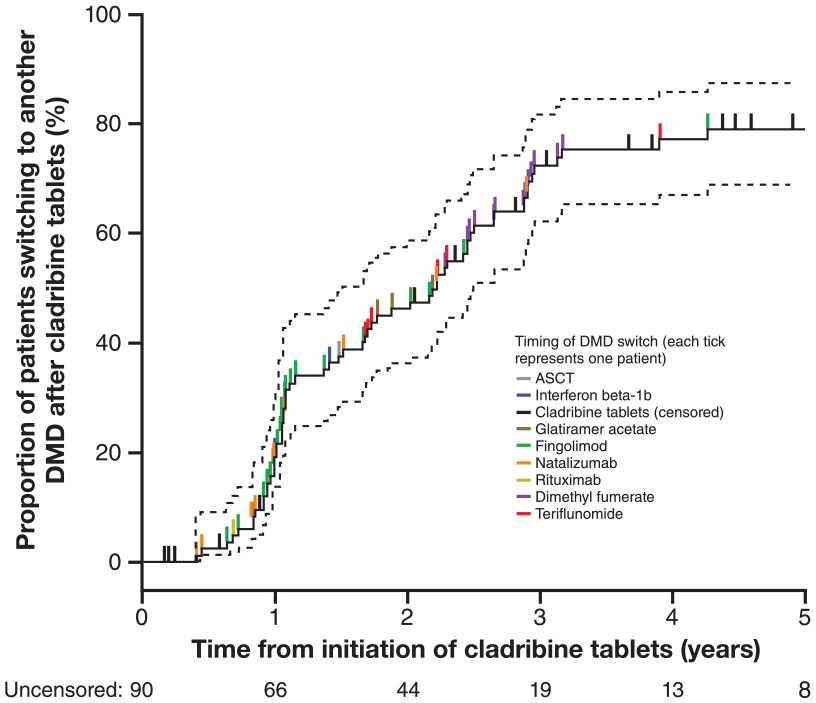
Patients treated with DMDs after cladribine treatment.
For the RRMS cohort, 47 of 70 patients (67%) received another DMD after treatment with cladribine during the follow-up period. Of the 47 patients who received further DMDs, 35 (74%) switched without having experienced a relapse (Table 3). The median time to next DMD for the RRMS cohort was 1.16 years (95% CI 1.06–1.79). The cumulative proportion of patients with RRMS commencing another therapy reached 23% at Year 1 after treatment with cladribine, 69% at Year 2 and 97% at Year 3.
Discussion
We retrospectively analysed records of patients in the Australian MSBase registry who received treatment with cladribine. The information in the MSBase database reflects clinical practice more closely than clinical trial populations, and the registry has a well-defined minimum dataset and publishes its data quality measures.12,16,17 Although patients were not able to receive the approved cumulative dose of cladribine (3.5 mg/kg over 2 years; instead only receiving 1.75 mg/kg in the first year of treatment), the dataset provides a follow-up of up to 5-year duration after treatment and complements the information available from clinical trials. The reconstructed disability trajectories suggest an overall increasing EDSS prior to treatment with cladribine. We observed that this trajectory was altered during the 2 years following treatment in RMS patients – a particularly notable finding given that this is a cohort of more disabled, older and extensively pre-treated RMS patients than patients in the clinical trials of cladribine tablets in MS. However, this observation should be interpreted with caution, as in an uncontrolled observational study any changes in disability trajectory after commencement of therapy may represent regression to the mean.
Specifically, in this cohort, mean age was 47 years and duration of MS was 13 years. In the CLARITY, CLARITY Extension, and ONWARD studies, the mean age at baseline was approximately 38 years.2,8,9 The mean age was even lower in the Phase 3 ORACLE-MS study of cladribine tablets in patients presenting with clinically isolated syndrome/first clinical demyelinating event, at approximately 32 years. 6 Increasing age is associated with an increased risk of disease progression, with a higher age and co-morbidities predicting worse disease outcomes.18–21 The patient population in this dataset also had a much higher EDSS (mean 4.7) at the commencement of cladribine compared with that of the populations randomised to cladribine at a cumulative dose of 3.5 mg/kg in CLARITY (mean 2.8), 2 ORACLE (mean 1.6–1.7), 5 ONWARD (mean 2.9), 9 and other recent trials. Despite their high age, long disease duration and high disability status at baseline, approximately 80% of patients remained free from EDSS progression and 65% were free from relapses 2 years after treatment with cladribine, consistent with observations in clinical trials. It could be hypothesised that patients who relapsed did experience further disability progression.
The efficacy of cladribine tablets appears to occur rapidly after administration of the first course, with effects on MRI activity evident at 3 months and effects on clinical parameters such as relapses and no evidence of disease activity (NEDA) evident at 6 months,22,23 and probably earlier as the relapse-free curves separate early. 2 Analysis of the dose of cladribine tablets suggests a lower efficacy over 2 years with doses lower than 3.5 mg/kg, but the effect of reduced doses on the duration of efficacy has not been formally examined. It is known that delaying the second-year course of cladribine by 6 months does not have a clinically meaningful impact on relapse risk over 2 years. 24
The lasting effect following the first-year dose may be explained by quantitative changes in key immune subsets that occur early in the treatment course and that are maintained at 1 year after dosing. Memory B cells, which are believed to play an important role in the pathophysiology of MS, are consistently reduced in number over a 12-month period, while immature B cell and transitional B cell counts recover at 12 months post-treatment.25–27 This is coupled with a more limited, but similarly long-term, depletion of CD4+ T cell subsets and to a lesser extent, CD8+ T cells. Consequently, any repopulating pathogenic cells emerge into a regulatory environment consisting of CD4+ T regulatory cells, CD8+ T suppressor cells and regulatory B cell subsets leading to the re-establishment of immune-tolerance that produces long-term control of MS.25–27 The quantitative, and perhaps qualitative, changes in key immune cells thought to be important in MS that persist for long after administration of cladribine tablets and long after the recovery of total lymphocyte counts combined with the sustained reduction of clinical activity seem to be an important feature of treatment with cladribine tablets.27–29
The therapeutic effect of cladribine appears to last for many months or years after the administration of the last dose in Year 2. In CLARITY Extension, clinical efficacy was maintained without further treatment with cladribine in Years 3 and 4. 4 A durable treatment effect on MRI outcomes was also seen for the majority of patients, with most patients remaining free of T1 gadolinium-enhancing lesions, even without further treatment in CLARITY Extension. 30 Only a small proportion of patients (approximately 10%) with adequate post-cladribine follow-up had MRI (brain or spinal) scans captured in the MSBase registry (data not shown). There is clinical interest in strategies for subsequent treatment with DMDs after cladribine treatment. Many of the patients in this cohort who received DMDs after cladribine received these treatments without progression or relapse. A reason for this may be that clinicians and patients made the decision to start another DMD as the full course of cladribine could not be completed because of regulatory restraints, rather than due to any clinical progression. This makes inference about the duration of clinical effect difficult to quantify in this study.
These data are also the first observations from clinical practice of patients with SPMS before, during, and after treatment with cladribine. The increasing EDSS trajectory in the 2 years prior to treatment with cladribine appeared to be ameliorated in the 2 years post-treatment in the patients with SPMS.
Some caveats should be noted relating to information in the MSBase registry. As with most observational registries, these caveats include a reasonable loss to follow-up, variable recording of outcomes over time among the participating centres, and relative scarcity of paraclinical data (notably, cerebral and spinal cord MRI descriptors). No comparison was made with other therapies, and there was no consistent follow-up for this cohort of patients, reflecting follow-up appointments and loss to follow-up or centre transfers that occur in routine clinical practice.
We focused on outcomes during the initial 2 years from treatment with cladribine, when information from 74% of the study cohort was available. Given the low patient numbers and observational nature of these data, no conclusions can be drawn on the effects of cladribine treatment in SPMS. Similarly, extrapolation of these results to the approved 2-year treatment schedule should be avoided as larger, representative post-marketing studies are needed.
Adverse event data are not systematically captured in the MSBase registry and were therefore not reported here. The adverse event profile for cladribine tablets has been reported from a pooled population of patients from the clinical development programme covering early to more advanced RMS. Lymphopenia was among the most frequently observed adverse events that occurred at a higher incidence with cladribine relative to placebo. There was no increased risk for infections in general during treatment with cladribine tablets except for a higher incidence of herpes zoster. 31
A recent real-life study in the United Kingdom reported on the off-label use of subcutaneous cladribine in a more disabled cohort of patients with relapsing and progressive MS who were not eligible for approved DMDs. Using a personalised dosing scheme to avoid the occurrence of severe lymphopenia, subcutaneous cladribine was shown to be well tolerated with few treatment-related adverse events; Grade 1 to 2 lymphopenia occurred in 50% of cases, a single patient developed transient Grade 3 lymphopenia, and no cases of varicella or other infections were observed. 32
Conclusion
Here, we summarise the clinical experience of patients treated with cladribine in the Australian cohort of the MSBase registry. These data were derived from patients who did not receive the full approved cumulative dose of cladribine, but the evidence is reflective of clinical practice prior to launch of the drug. The observational data reported here suggest a disease-modifying effect, in terms of reducing disability progression and relapses, in a patient cohort characterised by older age and more disability, but caution is needed in interpreting the clinical applicability from a single-arm cohort study. Further clinical trials and larger comparative post-marketing studies are needed to corroborate these findings.
Footnotes
Acknowledgements
This analysis was funded by a grant from Merck KGaA, Darmstadt, Germany, to MSBase. The analysis was performed by independent statisticians at the Clinical Outcomes Research unit at the University of Melbourne (T.K. and N.L.), and Merck KGaA had no access to the primary data. Medical writing support was provided by Ash Dunne and Elizabeth Strickland of inScience Communications and was funded by Merck KGaA, Darmstadt, Germany.
Declaration of Conflicting Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: This analysis was funded by Merck KGaA, Darmstadt, Germany. N.L. received conference travel support from Merck KGaA. S.H. received honoraria and consulting fees from Novartis, Bayer Schering, and Sanofi and travel grants from Novartis, Biogen Idec, and Bayer Schering. E.B. did not declare any competing interests. J.L.-S. accepted travel compensation from Novartis, Biogen, Roche, and Merck KGaA. Her institution received the honoraria for talks and advisory board commitment from Biogen, Sanofi-Genzyme, Merck KGaA, Novartis, and Teva; she has been involved in clinical trials with Biogen, Novartis, and Clene. M.S. has participated in, but not received honoraria for, advisory board activity for Biogen, Merck KGaA, Bayer Schering, Sanofi-Aventis, and Novartis. P.A.M. and C.S. did not declare any competing interests. O.S. received honoraria for advisory board commitment and talks as well as travel support from Roche, Sanofi-Genzyme, Merck KGaA, and Biogen. S.V. received honorarium from Merck KGaA. N.S. received honorarium for a medical advisory board for Merck KGaA. M.H.B. served on scientific advisory boards for Biogen, Novartis, and Sanofi-Genzyme and has received conference travel support from Biogen and Novartis. He serves on steering committees for trials conducted by Novartis. His institution has received research support from Biogen, Merck KGaA, and Novartis. J.P. served on scientific advisory boards for Biogen, Novartis, Roche, and Sanofi-Genzyme, accepted travel compensation from Novartis, Biogen, Sanofi-Genzyme, and Teva, and speaking honoraria from Biogen, Novartis, Sanofi-Genzyme, and Teva. H.B. institution (Monash University) has received compensation for consultancy, talks, advisory/steering committee activities from Biogen, Merck KGaA, Novartis, Genzyme, and Alfred Health; research support from Novartis, Biogen, Roche, Merck KGaA, NHMRC, Pennycook Foundation, and MSRA; and has received compensation for the same activities from Biogen, Merck KGaA, Oxford Health Policy Forum, and Novartis. D.J. is an employee of Merck KGaA, Darmstadt, Germany. J.F. is an employee of Merck Serono Australia Pty Ltd, a division of Merck KGaA, Darmstadt, Germany. T.K. served on scientific advisory boards for Roche, Sanofi-Genzyme, Novartis, Merck KGaA, and Biogen; received conference travel support and/or speaker honoraria from WebMD Global, Novartis, Biogen, Sanofi-Genzyme, Teva, bioCSL, and Merck; and has received research support from Biogen.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.