
Abstract
Background:
Diroximel fumarate (DRF) is a novel oral fumarate for patients with relapsing–remitting multiple sclerosis (RRMS). DRF and the approved drug dimethyl fumarate yield bioequivalent exposure to the active metabolite monomethyl fumarate; thus, efficacy/safety profiles are expected to be similar. However, DRF’s distinct chemical structure may result in a differentiated gastrointestinal (GI) tolerability profile.
Objective:
To report interim safety/efficacy findings from patients in the ongoing EVOLVE-MS-1 study.
Methods:
EVOLVE-MS-1 is an ongoing, open-label, 96-week, phase 3 study assessing DRF safety, tolerability, and efficacy in RRMS patients. Primary endpoint is safety and tolerability; efficacy endpoints are exploratory.
Results:
As of March 2018, 696 patients were enrolled; median exposure was 59.9 (range: 0.1–98.9) weeks. Adverse events (AEs) occurred in 84.6% (589/696) of patients; the majority were mild (31.2%; 217/696) or moderate (46.8%; 326/696) in severity. Overall treatment discontinuation was 14.9%; 6.3% due to AEs and <1% due to GI AEs. At Week 48, mean number of gadolinium-enhancing lesions was significantly reduced from baseline (77%; p < 0.0001) and adjusted annualized relapse rate was low (0.16; 95% confidence interval: 0.13–0.20).
Conclusion:
Interim data from EVOLVE-MS-1 suggest DRF is a well-tolerated treatment with a favorable safety/efficacy profile for patients with RRMS.
Keywords
Introduction
Diroximel fumarate (DRF) is a novel oral fumarate in development for patients with relapsing–remitting multiple sclerosis (RRMS). DRF undergoes rapid esterase cleavage in the gut to monomethyl fumarate (MMF), the same pharmacologically active metabolite as the approved drug delayed-release dimethyl fumarate (DMF). 1 DMF has demonstrated significant and clinically meaningful efficacy in clinical trials and real-world studies totaling >780,000 patient-years of exposure.2–6 At the investigational dose of 462 mg, DRF results in MMF systemic exposure that is bioequivalent to DMF 240 mg (Supplemental Figure 1). 7 Therefore, efficacy and safety profiles for DRF and DMF are expected to be similar.
DRF is differentiated from DMF based on its distinct chemical structure (Figure 1). DRF is hypothesized to produce less irritation and reactivity toward off-target receptors within the gastrointestinal (GI) tract than DMF, potentially leading to improved GI tolerability. 8 A secondary major metabolite of DRF hydrolysis is 2-hydroxyethyl succinimide (HES). HES has been studied extensively in preclinical, in vitro, and healthy volunteer studies. No evidence of potential pharmacological activity or toxicology findings related to HES have been demonstrated.
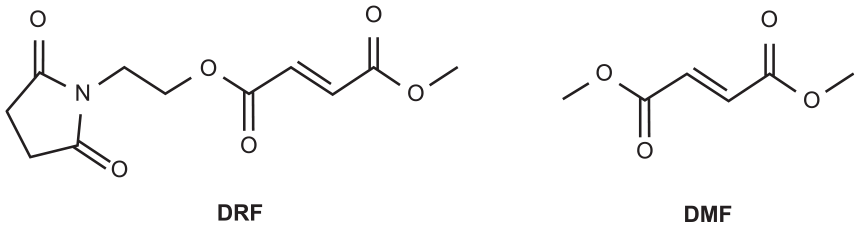
Chemical structures of DRF and DMF.
EVOLVE-MS-1 is an ongoing phase 3 study to evaluate DRF in adults with RRMS, undertaken to ensure that there were no unexpected safety events in a large patient cohort followed prospectively over 2 years. Here, we report interim results from a 30 March 2018 data cut, comprising 696 enrolled patients with a median DRF exposure of approximately 1 year, representing 685 patient-years of exposure.
Methods
Study design
EVOLVE-MS-1 (NCT02634307) is an ongoing, open-label, single-arm, phase 3 study assessing long-term safety, tolerability, and efficacy of DRF 462 mg twice daily over 96 weeks in patients with RRMS. The study consists of 4-week screening, 96-week treatment, and 2-week safety follow-up (Figure 2). Approximately 800–1000 patients are planned for this study. The study population includes (1) patients who rolled over from the 5-week, randomized, double-blind, phase 3 EVOLVE-MS-2 (NCT03093324) study of DRF and DMF, and (2) those newly enrolled to the DRF clinical trial program. DRF 462 mg was administered via oral capsules twice daily; a 1-week titration schedule was used for patients receiving DRF for the first time (Figure 2).
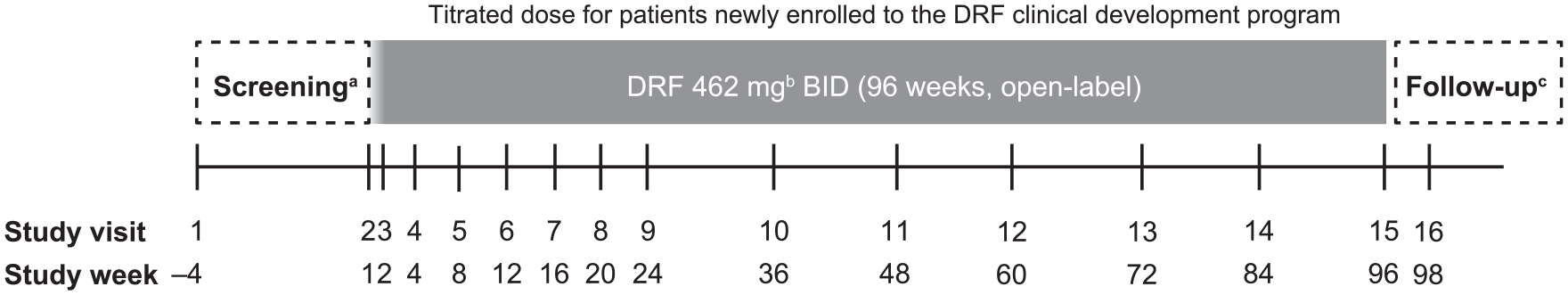
EVOLVE-MS-1 study design.
Patients
Eligible patients aged 18–65 years had a confirmed diagnosis of RRMS 9 and were neurologically stable with no evidence of relapse in the 30 days before screening. Prior disease-modifying therapy (DMT), including DMF, was permitted. Complete inclusion and exclusion criteria are described in the Supplementary Appendix. The study was approved by central and local ethics committees and conducted in accordance with the International Council on Harmonization guidelines for Good Clinical Practice and the Declaration of Helsinki. All patients provided written, informed consent.
Study endpoints
The primary endpoint was DRF safety and tolerability. Exploratory efficacy endpoints included radiological (gadolinium-enhancing (Gd+), new/newly enlarging T2, new T1 hypointense lesion counts), clinical (annualized relapse rate (ARR), MS relapse, Expanded Disability Status Scale (EDSS) score, no evidence of disease activity-3 (NEDA-3), timed 25-foot walk (T25-FW) score), and patient-reported outcomes ((PROs); EuroQol Group Heath Outcome Measure 5 Level (EQ-5D-5L) and 12-Item Short Form Health Survey (SF-12)).
Assessments
Safety evaluations included treatment-emergent adverse events (TEAEs) and laboratory parameters (chemistry, hematology, and urinalysis). Absolute lymphocyte count (ALC) lower limit of normal (LLN) was defined as <0.91 × 109/L. DRF was temporarily withheld if ALC reached a confirmed level of <0.5 × 109/L and permanently discontinued if levels remained <0.5 × 109/L for ⩾4 weeks, or per investigator discretion. Patients who permanently discontinued the study with a last measured ALC of <0.8 × 109/L were followed for 6 months afterward for lymphocyte monitoring. Moderate and severe prolonged lymphopenia were defined as ALC <0.8–0.5 × 109/L and <0.5 × 109/L, respectively, sustained for >6 months.
Tolerability-related adverse events (AEs) were classified using the Medical Dictionary for Regulatory Activities Preferred Terms within the System Organ Class for gastrointestinal (GI) disorders and for flushing/flushing-related AEs.
Protocol-defined MS relapse was defined as new or recurrent neurologic symptoms (not associated with fever/infection) lasting ⩾24 hours and accompanied by new neurological findings and change in EDSS score. ARR values on study reflect protocol-defined MS relapses. Baseline ARR values reflect patient-reported relapses in the 12 months before study start. NEDA-3 was defined as no relapses, no confirmed disability progression sustained for 12 weeks per EDSS, and no new/newly enlarging T2 hyperintense or Gd+ lesions. Written documentation was required to confirm a patient’s willingness to continue the study in the instance of MS relapse, disability progression measured by EDSS, or total Gd+ lesion count ⩾5 assessed by the central magnetic resonance imaging (MRI) facility.
Analysis populations and statistics
This data cut was preplanned for early 2018 and targeted because a cohort of patients would have meaningful exposure to study drug. Relapses and AEs were assessed in the overall (intention-to-treat) population, defined as all patients who received ⩾1 DRF dose in EVOLVE-MS-1. Safety events and laboratory changes that may occur early in therapy, such as changes in ALC or onset of GI and flushing events, were also evaluated in patients with no prior fumarate exposure (fumarate-naïve subgroup). Radiological and disability assessments were performed in patients who completed ⩾1 post-baseline efficacy assessment. Efficacy endpoints also were evaluated in patients who were diagnosed with RRMS within 1 year of study entry and naïve to DMTs (newly diagnosed subgroup).
Summary statistics were provided for all parameters. MRI parameters and protocol-defined MS relapse were summarized using descriptive statistics. Adjusted ARR was based on a Poisson regression model. Baseline was defined as on/after first dose in EVOLVE-MS-1 for AEs and on/before first dose in EVOLVE-MS-1 for laboratory/efficacy assessments; for EVOLVE-MS-2 rollover patients, baseline disease characteristics were collected from the EVOLVE-MS-2 baseline visit.
Results
Patients
As of 30 March 2018, 696 patients (including 556 fumarate-naïve patients) received ⩾1 DRF dose. Mean patient age in the overall population was 42 years; 65% received prior DMTs, and 41% were enrolled in US sites, with the remaining in Europe and Canada (Table 1). At cutoff, 83.2% of patients were on treatment, 1.9% completed the study, and 14.9% discontinued treatment. Median (range) DRF exposure was 59.9 (0.1–98.9) weeks; mean (standard deviation (SD)) exposure was 51.3 (23.9) weeks.
Baseline demographics and disease characteristics in EVOLVE-MS-1.
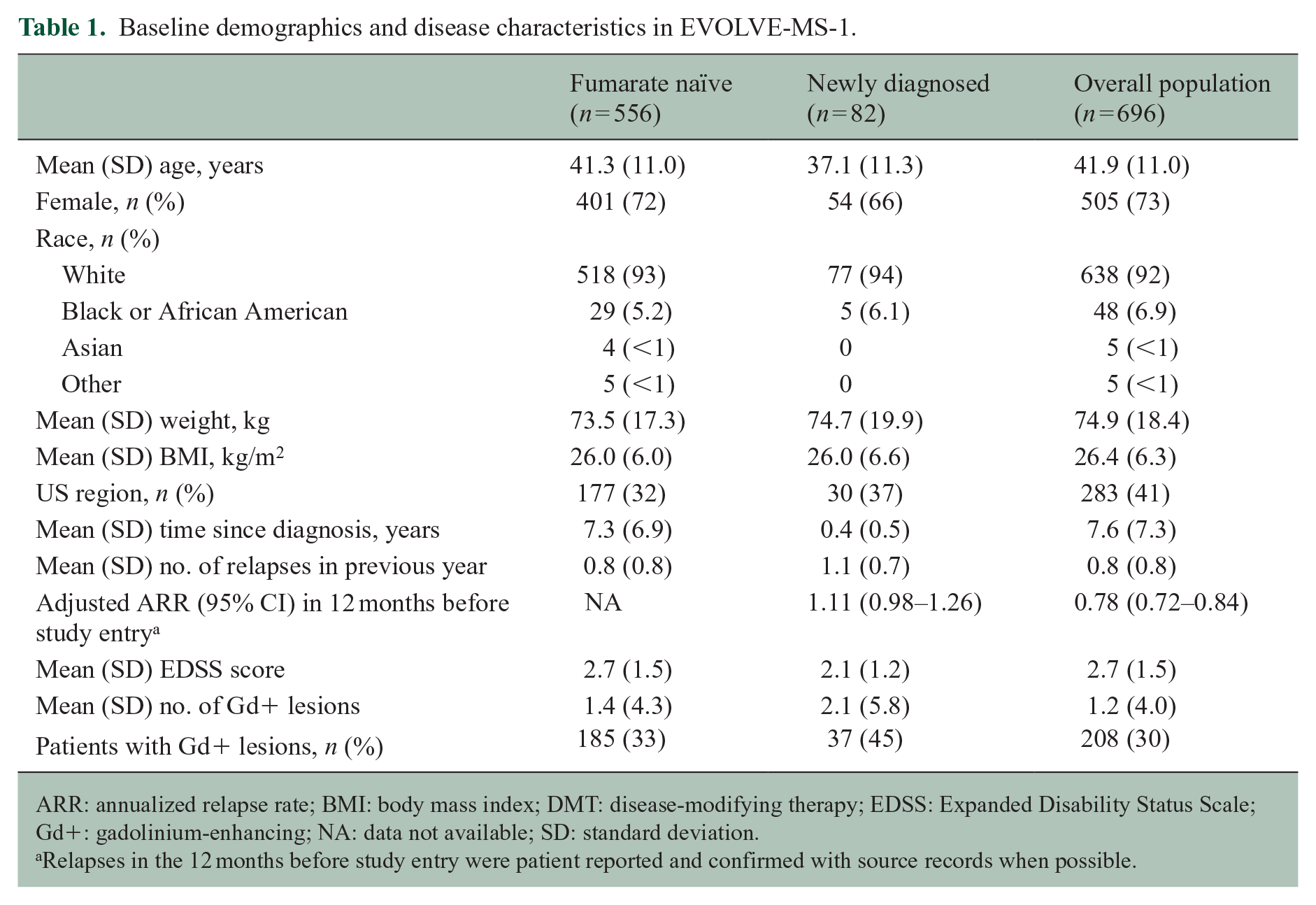
ARR: annualized relapse rate; BMI: body mass index; DMT: disease-modifying therapy; EDSS: Expanded Disability Status Scale; Gd+: gadolinium-enhancing; NA: data not available; SD: standard deviation.
Relapses in the 12 months before study entry were patient reported and confirmed with source records when possible.
Safety
Summary of safety during the treatment period
TEAEs were reported in 84.6% (589/696) of the overall population (Table 2). In most patients, TEAEs were mild (31.2%; 217/696) or moderate (46.8%; 326/696) in severity. The most common TEAE was flushing (34.1%); TEAEs occurring in ⩾10% patients are shown in Table 2. Serious AEs were reported in 7.5% (52/696). Two deaths (0.3%) occurred; one because of an accidental fall and one because of a hypertensive cardiovascular event. Both were deemed unrelated to study drug by the investigator.
Summary of safety during the treatment period.
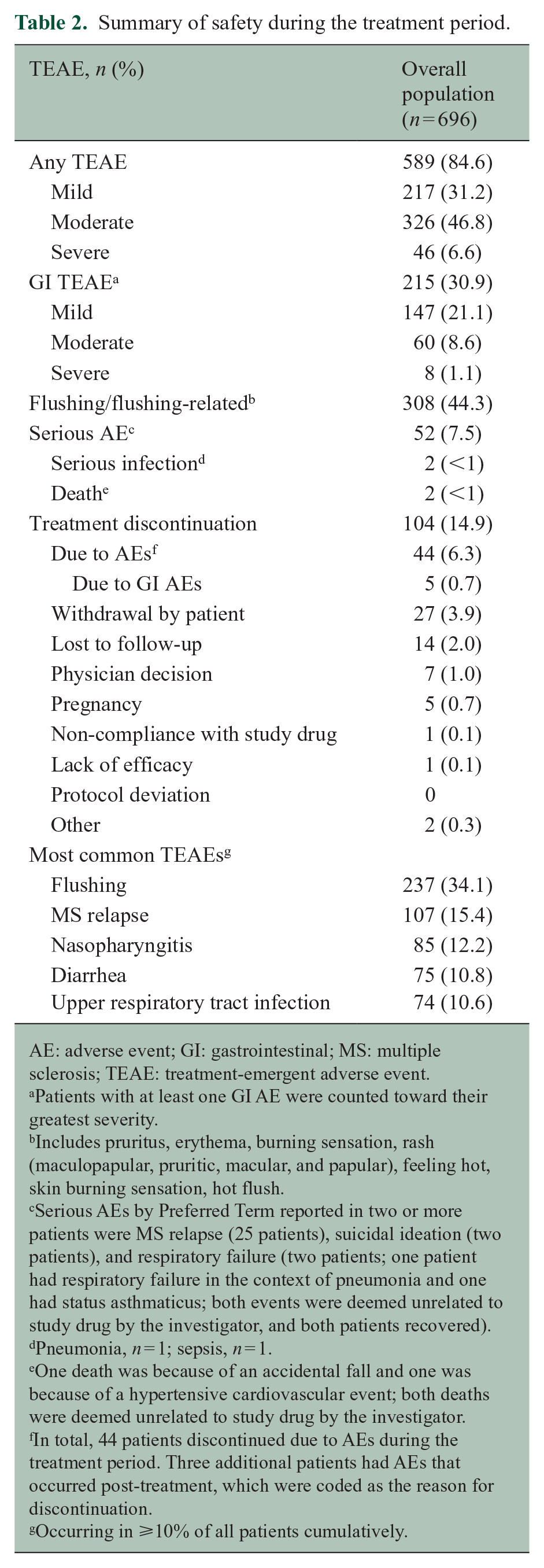
AE: adverse event; GI: gastrointestinal; MS: multiple sclerosis; TEAE: treatment-emergent adverse event.
Patients with at least one GI AE were counted toward their greatest severity.
Includes pruritus, erythema, burning sensation, rash (maculopapular, pruritic, macular, and papular), feeling hot, skin burning sensation, hot flush.
Serious AEs by Preferred Term reported in two or more patients were MS relapse (25 patients), suicidal ideation (two patients), and respiratory failure (two patients; one patient had respiratory failure in the context of pneumonia and one had status asthmaticus; both events were deemed unrelated to study drug by the investigator, and both patients recovered).
Pneumonia, n = 1; sepsis, n = 1.
One death was because of an accidental fall and one was because of a hypertensive cardiovascular event; both deaths were deemed unrelated to study drug by the investigator.
In total, 44 patients discontinued due to AEs during the treatment period. Three additional patients had AEs that occurred post-treatment, which were coded as the reason for discontinuation.
Occurring in ⩾10% of all patients cumulatively.
Tolerability
GI TEAEs occurred in 30.9% (215/696) of the overall population, with similar incidence in fumarate-naïve patients (31.3%; 174/556). Of patients with a GI event (215/696), the majority of events were mild (68%; 147/215) or moderate (28%; 60/215) in severity; 84/215 (39%) patients received transient concomitant therapy for GI symptoms. GI TEAEs resolved in 89% (191/215): median (10th–90th percentile) duration was 7.5 (1–87) days. Of patients with GI TEAEs with complete start dates recorded (n = 214), most (60%; 129/214) had events within the first month of treatment (Figure 3(a)). Serious GI TEAEs occurred in three (0.4%) patients (1 patient each of abdominal pain, inguinal hernia, peptic ulcer).
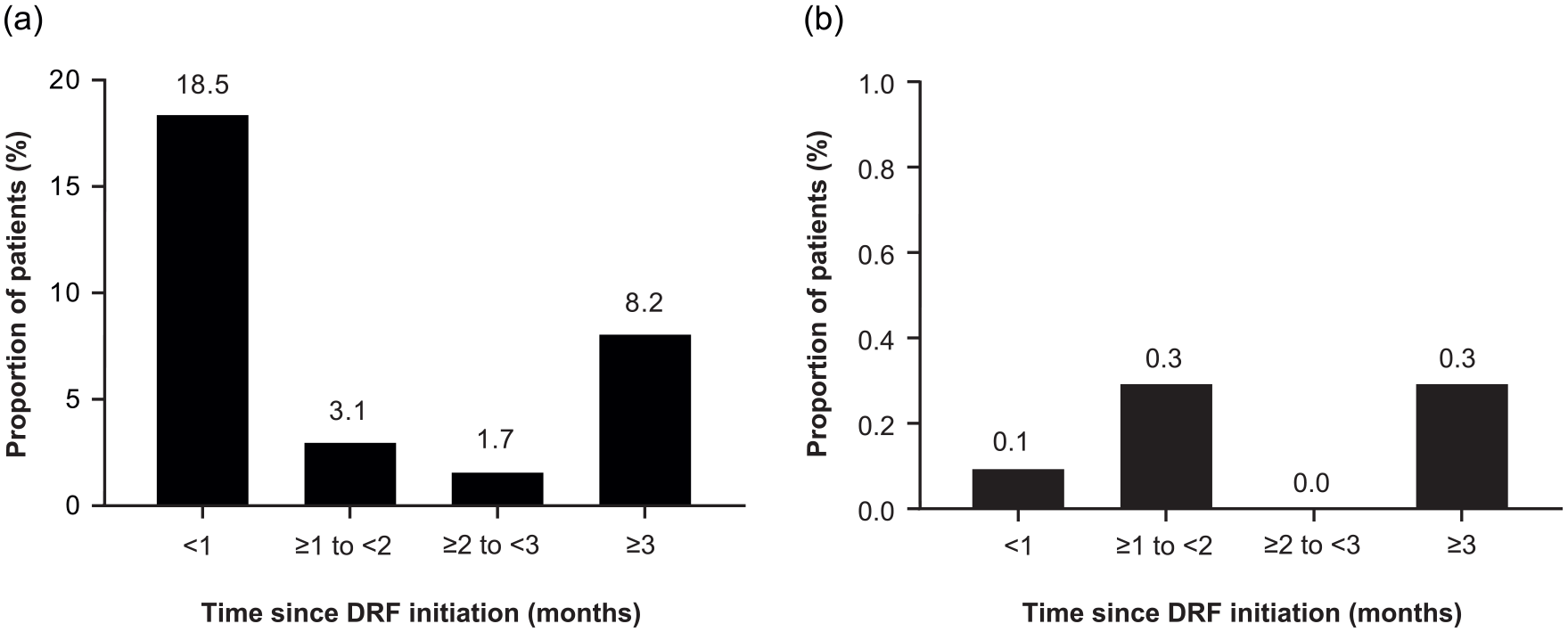
Onset of GI AEs and treatment discontinuation due to GI AEs by time interval in EVOLVE-MS-1. (a) Proportion of patients experiencing onset of first GI AE was reported by time since DRF initiation in the overall population. (b) Proportion of patients experiencing DRF treatment discontinuations due to GI AEs was reported by time since DRF initiation in the overall population.
Overall, flushing/flushing-related TEAEs were reported in 44.3% (308/696), with generally similar incidence in fumarate-naïve patients (47.3%; 263/556). In patients with flushing/flushing-related events, most were mild (79%; 244/308) or moderate (19%; 60/308). Among patients with flushing/flushing-related events with complete start dates, the majority (80.3%; 245/305) had events within the first month of treatment; median (25th–75th percentile) time to onset for first event was 1 (1–18) day. Flushing/flushing-related events resolved in 74% (229/308): median (10th–90th percentile) duration was 3.5 (1–88) days.
DRF treatment was prematurely discontinued by 14.9% (104/696) of patients. AEs were the most common reason for discontinuation in the overall population (6.3%; 44/696) and fumarate-naïve subgroup (6.8%; 38/556) overall, 0.7% (5/696) discontinued DRF because of GI AEs; four of the five were fumarate naïve (4/556), and the majority occurred early in treatment (<2 months; Figure 3(b)). AEs leading to treatment discontinuation are shown in Supplemental Table 1.
Lymphocyte counts
ALCs declined by approximately 28.4% within the first year and then stabilized, remaining more than LLN for the majority of patients (64.8%; 441/681; Figure 4). A similar magnitude and pattern of decline followed by stabilization was observed in fumarate-naïve patients. Incidence of prolonged moderate lymphopenia (<0.8–0.5 × 109/L sustained for 6 months) was 7.3% (50/681). No patients developed prolonged (⩾6 months) severe (0.5 × 109/L) lymphopenia, as patients were required to discontinue DRF treatment if ALCs were <0.5 × 109/L for ⩾4 weeks (protocol-defined stopping rule).
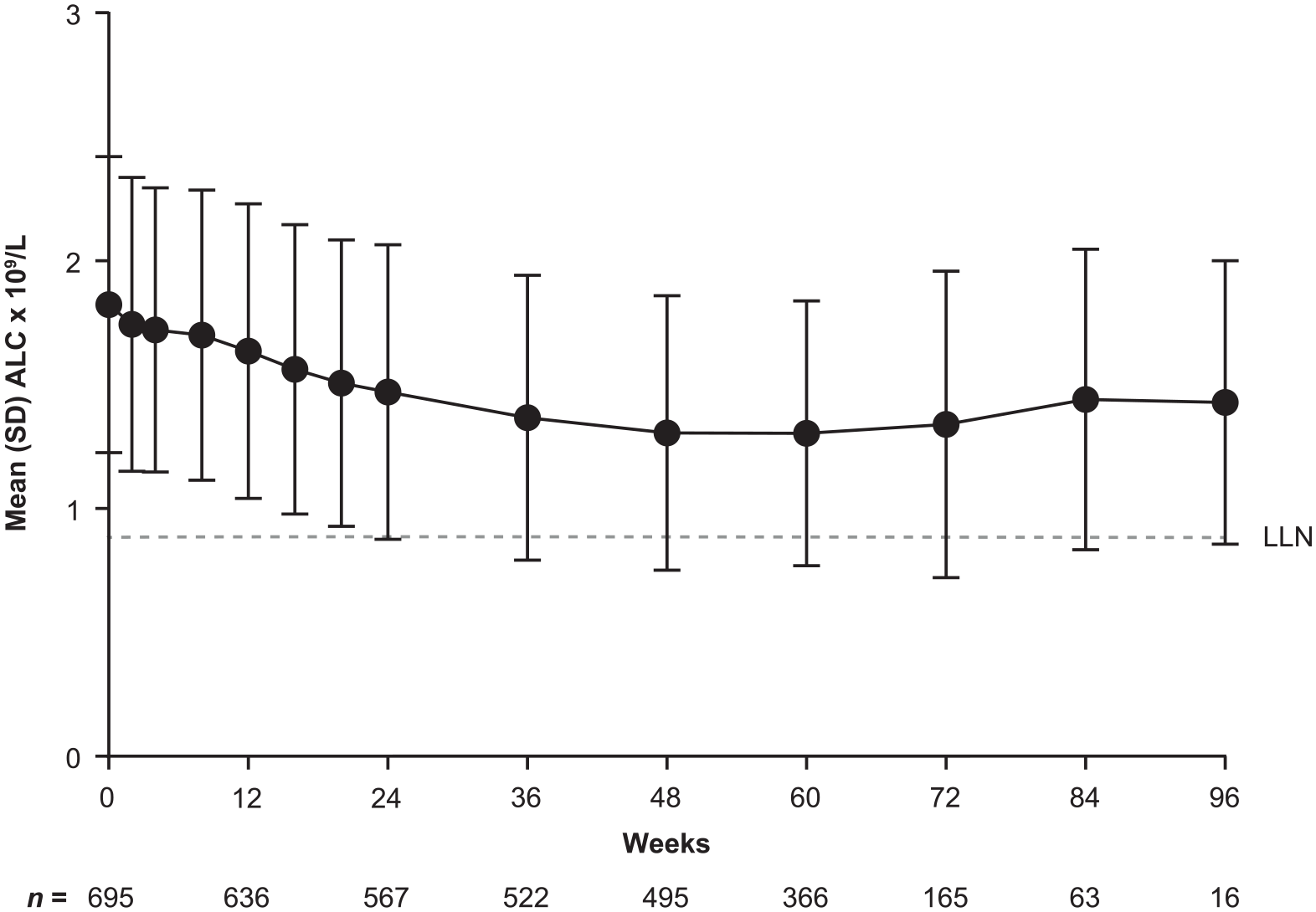
ALCs over time in DRF-treated patients in EVOLVE-MS-1.
To date, six patients had an ALC less than LLN at the time of DRF discontinuation and remained on study for lymphocyte reconstitution assessment. Four of the six patients reached LLN during the follow-up period; median time to reach LLN after discontinuation was 5.0 (range, 4.0–7.4) months. All four of these patients had at least two consecutive ALCs <0.5 × 109/L while on treatment. The two patients who did not reach LLN have <3 months of follow-up after discontinuation and remain on study for continued assessment (Supplemental Figure 2).
AEs of special interest
Two (0.3%) serious infections were reported (pneumonia, n = 1; sepsis, n = 1), neither in the context of severe prolonged lymphopenia. There were no serious opportunistic infections. Malignancy occurred in two (0.3%) patients (invasive ductal breast carcinoma, n = 1; cervical carcinoma, n = 1). There were no observed cases of anaphylaxis or angioedema.
Elevations in serum alanine aminotransferase (ALT) and aspartate aminotransferase (AST) >3×, >5×, and >10× upper limit of normal (ULN) occurred in 16 (2.3%), 7 (1.0%), and 2 (0.3%) patients, and 9 (1.3%), 5 (0.7%), and 2 (0.3%) patients, respectively. No cases met Hy’s law criteria (i.e. concurrent elevation of total bilirubin >2× ULN). ALT and AST elevations resolved in 88% (14/16) and 89% (8/9) of patients, respectively; median (range) time to resolution was 11.5 (4–144) and 13 (1–113) days. Four (0.6%) patients discontinued treatment because of increased liver function tests. No cases of irreversible liver injury or liver failure have been reported to date.
Renal injury TEAEs occurred in 1.6% (11/696) of patients; all were mild or moderate in severity (none were serious or led to treatment discontinuation). The most common event was proteinuria (0.6%; 4/696).
Efficacy
Clinical endpoints
Overall, adjusted ARR was 0.16 (95% confidence interval (CI): 0.13–0.20) at Week 48. A similar adjusted ARR after 1 year of treatment (0.19; 95% CI: 0.10–0.33) was observed in newly diagnosed patients (Figure 5). Estimated proportion of patients with MS relapse at Week 48 was similar for both groups (overall, 13.1%; newly diagnosed, 14.0%). By Week 48, 88.8% of the overall population had no relapses, 9.5% had one relapse, 1.6% had two, none had three, and 0.1% had four or more. Mean (SD) EDSS score at baseline and Week 48 was 2.70 (1.48) and 2.75 (1.50) overall, and 2.10 (1.15) and 2.25 (1.10) in newly diagnosed patients.
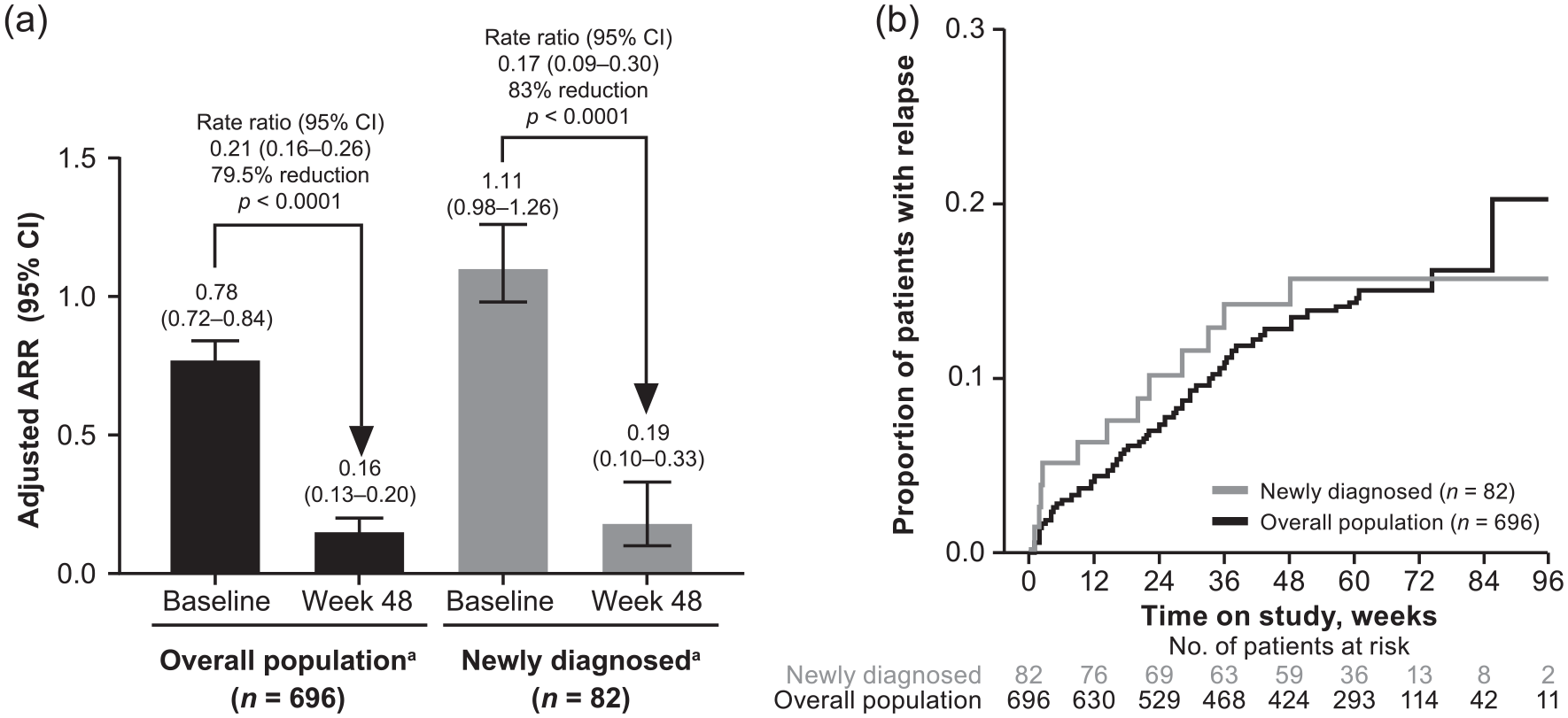
ARR and proportion with relapse at Week 48 in EVOLVE-MS-1. (a) Adjusted ARR (95% CI) was calculated in the overall population and newly diagnosed patients based on a Poisson regression model. Week 48 values represent the number of protocol-defined relapses occurring on or before 48 weeks. Baseline values represent the 12 months before DRF initiation and are patient reported. (b) Estimated proportion of patients with protocol-defined relapse at Week 48 in the overall population and newly diagnosed patients.
Radiological endpoints
Mean Gd+ lesion count was reduced from baseline to Week 48 by 77% (p < 0.0001) in patients with Week 48 MRI assessments (n = 503) and by 96% (p = 0.0051) in newly diagnosed patients (n = 70; Figure 6(a)). A greater percentage of patients in both populations had zero Gd+ lesions at Week 48 compared with baseline (Figure 6(b)). Mean new/newly enlarging T2 and new T1 hypointense lesion counts from baseline to Week 48 were similar for both populations (Figure 6(c) and (d)). Estimated proportion of evaluable patients (n = 508) with NEDA-3 at Week 48 was 38.1%.
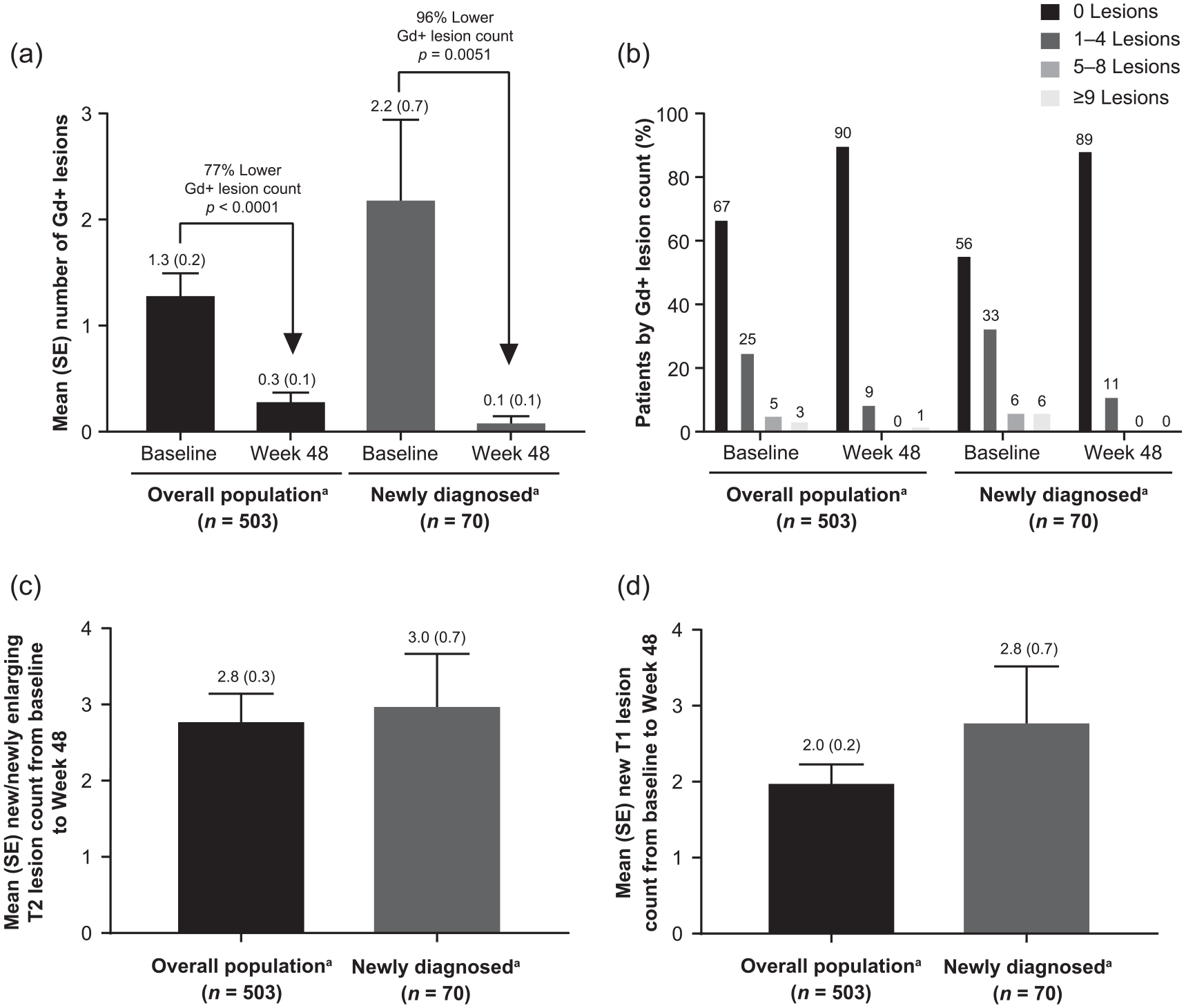
MRI lesions from baseline to Week 48 in EVOLVE-MS-1. (a) Mean number of Gd+ lesions, (b) Gd+ lesions by numerical category, (c) new/newly enlarging T2 hyperintense lesions, and (d) new T1 hypointense lesions evaluated from baseline to Week 48 in the overall population and newly diagnosed patients in EVOLVE-MS-1.
T25-FW scores and PROs
T25-FW scores and PROs (SF-12 and EQ-5D-5L) generally remained stable over the first year of treatment (Supplementary Material). However, longer follow-up is warranted.
Discussion
DRF is a novel oral fumarate in development for RRMS with a chemical structure that is theorized to be less GI-irritating than DMF. Safety and efficacy profiles for DRF are expected to be similar to DMF based on bioequivalent systemic exposure to MMF when administered at the investigated and approved doses, respectively. Although EVOLVE-MS-1 did not include a comparator arm, these interim findings demonstrate a safety profile consistent with the known DMF clinical trial and real-world experience, which has been well characterized in over 780,000 patient-years of exposure.2–4,6,10 Thus far, DRF safety events are consistent with DMF, and laboratory changes and tolerability events occur over a similar time-course.2,3,10,11
Lymphocyte decline over the first year of treatment followed by stabilization is a known phenomenon with DMF. 10 As expected, DRF demonstrated a similar impact on ALC with respect to overall magnitude and pattern of decline. In the small subset of DRF-treated patients who developed prolonged lymphopenia, the pattern of ALC decline was similar to the overall population whose ALCs remained more than LLN, and the incidence was similar to the DMF experience. This supports ALC monitoring as an effective tool for identifying patients at risk of developing severe prolonged lymphopenia, a risk factor for DMF-associated progressive multifocal leukoencephalopathy (PML).1,4 Only six patients discontinued DRF with lymphopenia and are being assessed for post-DRF lymphocyte reconstitution; this population will be explored further as patient numbers increase. To date, no serious opportunistic infections, including PML, have been reported.
Transient increases in liver transaminases (typically ⩽3× ULN) were observed with treatment initiation, with slightly greater mean increases from baseline in ALT than AST. Levels subsided with continued treatment, consistent with DMF. 1 There were no clinically meaningful or numerically significant mean changes from baseline observed for ALT, AST, alkaline phosphatase, or bilirubin. No liver function test abnormalities fulfilled Hy’s law criteria, and incidence of renal TEAEs was low. Analysis of renal and hepatic events provides no evidence of renal or hepatic injury associated with DRF.
GI events, typically occurring within 1–2 months of treatment initiation, are among the most commonly reported AEs in clinical and real-world studies of DMF and have led to DMF treatment discontinuation in up to 20% of patients.2,3,11–16 In EVOLVE-MS-1, DRF appeared to be associated with lower than expected rates of GI and serious GI events based on studies with other fumaric acid esters.1,17 In DRF-treated patients with GI AEs, events typically occurred within the first month of treatment and were short, lasting 7.5 days for most patients. GI AEs were generally mild-to-moderate in severity and led to discontinuation in five (0.7%) patients to date. Overall treatment discontinuation rates appeared low at 14.9%, with 6.3% due to AEs. For reference, in Year 1 of the DEFINE 3 and CONFIRM 2 trials, GI AEs occurred in 36% of patients, and rates of discontinuation due to AEs and GI AEs were 11% and 3%, respectively, with DMF 240 mg twice daily (Biogen, unpublished data). Although firm conclusions cannot be made regarding GI tolerability compared with DMF, the low rates of GI events and GI discontinuations in EVOLVE-MS-1 suggest that DRF is well-tolerated. The EVOLVE-MS-2 head-to-head study of DRF versus DMF will directly assess tolerability differences.
Adjusted ARR at Week 48 was 0.16 (95% CI: 0.13–0.20), which was similar to that observed with DMF in the open-label ESTEEM trial (0.18; 95% CI: 0.15–0.20; n = 2011). 18 Compared with the patient-reported adjusted ARR for the 12 months before study entry (0.78; 95% CI: 0.72–0.84); the rate ratio was 0.21 (95% CI: 0.16–0.26; p < 0.0001), representing a 79.5% reduction from baseline. It should be noted, however, that the relapse rate on study reflects protocol-defined, investigator-confirmed relapses, whereas the baseline rate reflects patient-reported relapses that were verified with source records when possible. Reductions from baseline in Gd+ lesion count in the overall cohort and newly diagnosed patients supports the effectiveness of DRF across the spectrum of MS patients included in this study. Efficacy in newly diagnosed patients is clinically meaningful because early control of disease activity is associated with better outcomes. 19 Although efficacy endpoints are exploratory and EVOLVE-MS-1 is an open-label study, reductions in ARR and MRI outcomes at 1 year support the underlying assumptions regarding MMF bioequivalence and expected similar efficacy. A direct comparison of efficacy outcomes from EVOLVE-MS-1 and DMF phase 3 trials using propensity score-matching to appropriately adjust for differences in trial design and baseline characteristics may be valuable.
Notably, EVOLVE-MS-1 is an open-label study with no comparator arm and therefore unable to demonstrate the efficacy of DRF. However, given that DRF 462 mg and DMF 240 mg produce bioequivalent exposures of MMF, no differences in the efficacy profiles of DRF and DMF are expected. Second, tolerability could be over-reported if patients are aware that GI events are being assessed, or under-reported if they perceive treatment benefit. Therefore, it will be critical to fully describe any tolerability benefit in a setting with a proper control arm or a model using clinical trials mimicking these conditions. Real-world tolerability experience, which may differ from the clinical trial setting, will also continue to inform the DRF tolerability profile. Interestingly, tolerability events and laboratory changes expected to occur early in treatment occurred at similar rates for patients in the overall and fumarate-naïve population. Given the relatively small number of EVOLVE-MS-2 rollover patients (15%; 103/696), the short study duration (i.e. 4 weeks of maintenance dose), and that GI events typically occur in the first 1–2 months of therapy, firm conclusions cannot be made on whether tolerability differences exist in patients recently treated with fumarates versus those who are not. In addition, duration of follow-up for this interim analysis (median exposure ~1 year) limits the ability to assess some potential long-term safety and efficacy outcomes (e.g. malignancy, opportunistic infection, and disability progression). However, thus far there have been no unexpected safety or efficacy findings with DRF compared with the 2-year clinical trial DMF experience, which has been consistent in long-term (up to 12 years’ exposure) extension studies and real-world experience.2–4,6,20
Conclusion
DRF is a novel oral fumarate with a distinct chemical structure in development for patients with RRMS. Interim findings from EVOLVE-MS-1 suggest DRF has a favorable safety and efficacy profile, and appears to be a well-tolerated treatment option.
Supplemental Material
MSJ-19-0334.R2_Supplemental_Figure_1 – Supplemental material for Diroximel fumarate (DRF) in patients with relapsing–remitting multiple sclerosis: Interim safety and efficacy results from the phase 3 EVOLVE-MS-1 study
Supplemental material, MSJ-19-0334.R2_Supplemental_Figure_1 for Diroximel fumarate (DRF) in patients with relapsing–remitting multiple sclerosis: Interim safety and efficacy results from the phase 3 EVOLVE-MS-1 study by Robert T Naismith, Jerry S Wolinsky, Annette Wundes, Christopher LaGanke, Douglas L Arnold, Dragana Obradovic, Mark S Freedman, Mark Gudesblatt, Tjalf Ziemssen, Boris Kandinov, Ilda Bidollari, Maria Lopez-Bresnahan, Narinder Nangia, David Rezendes, Lili Yang, Hailu Chen, Shifang Liu, Jerome Hanna, Catherine Miller and Richard Leigh-Pemberton in Multiple Sclerosis Journal
Supplemental Material
MSJ-19-0334.R2_Supplemental_Figure_2 – Supplemental material for Diroximel fumarate (DRF) in patients with relapsing–remitting multiple sclerosis: Interim safety and efficacy results from the phase 3 EVOLVE-MS-1 study
Supplemental material, MSJ-19-0334.R2_Supplemental_Figure_2 for Diroximel fumarate (DRF) in patients with relapsing–remitting multiple sclerosis: Interim safety and efficacy results from the phase 3 EVOLVE-MS-1 study by Robert T Naismith, Jerry S Wolinsky, Annette Wundes, Christopher LaGanke, Douglas L Arnold, Dragana Obradovic, Mark S Freedman, Mark Gudesblatt, Tjalf Ziemssen, Boris Kandinov, Ilda Bidollari, Maria Lopez-Bresnahan, Narinder Nangia, David Rezendes, Lili Yang, Hailu Chen, Shifang Liu, Jerome Hanna, Catherine Miller and Richard Leigh-Pemberton in Multiple Sclerosis Journal
Supplemental Material
MSJ881761_supplementary_appendix – Supplemental material for Diroximel fumarate (DRF) in patients with relapsing–remitting multiple sclerosis: Interim safety and efficacy results from the phase 3 EVOLVE-MS-1 study
Supplemental material, MSJ881761_supplementary_appendix for Diroximel fumarate (DRF) in patients with relapsing–remitting multiple sclerosis: Interim safety and efficacy results from the phase 3 EVOLVE-MS-1 study by Robert T Naismith, Jerry S Wolinsky, Annette Wundes, Christopher LaGanke, Douglas L Arnold, Dragana Obradovic, Mark S Freedman, Mark Gudesblatt, Tjalf Ziemssen, Boris Kandinov, Ilda Bidollari, Maria Lopez-Bresnahan, Narinder Nangia, David Rezendes, Lili Yang, Hailu Chen, Shifang Liu, Jerome Hanna, Catherine Miller and Richard Leigh-Pemberton in Multiple Sclerosis Journal
Footnotes
Acknowledgements
The authors thank the EVOLVE-MS-1 study patients, investigators, and staff. Biogen provided funding for medical writing support in the development of this manuscript; Susan Chow, PhD, from Excel Scientific Solutions wrote the first draft of the manuscript based on input from authors, and Miranda Dixon from Excel Scientific Solutions copyedited and styled the manuscript per journal requirements. The authors had full editorial control of the manuscript and provided their final approval of all content.
Declaration of Conflicting Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: R.T.N. reports compensation as an advisor, consultant, or speaker for Alexion, Alkermes, Biogen, Celgene, EMD Serono, Genentech, Genzyme, Novartis, TG Therapeutics, and Viela Bio. J.S.W. reports compensation for consulting, scientific advisory boards, or other activities with Alkermes, AbbVie, Actelion, Acorda, Bayer, Celgene, Forward Pharma, EMD Serono, GeNeuro, GW Pharma, MedDay, Novartis, Otsuka, PTC, Roche/Genentech, Sanofi-Genzyme, Takeda, and Teva, and royalties for out-licensed monoclonal antibodies through UTHealth from Millipore Corporation. A.W. reports research support from Alkermes, AbbVie, and Biogen, and compensation as an advisor from Biogen. C.L. reports advisory, consultancy, and speaker activities for Acorda, Bayer, Biogen, EMD Serono, Genzyme, Novartis, Pfizer, Questcor, and Teva, and research support from Bayer, Biogen, Genzyme, GlaxoSmithKline, Novartis, Pfizer, Teva, and Vaccinex. D.L.A. reports honoraria from Biogen, Celgene, GeNeuro, Genentech, Roche, Merck, Novartis, Sanofi, Teva, and Wave Life Sciences; research support from Biogen, Immunotec, and Novartis; and an equity interest in NeuroRx. D.O. reports advisory, consultancy, and speaker activities for Genzyme, Merck, Roche, and Teva. M.S.F. reports research or educational grants from Genzyme Canada; honoraria or consultation fees from Actelion, Bayer, Biogen, Celgene, Chugai, EMD Canada, Genzyme, Merck Serono, Novartis, Hoffman La-Roche, PENDOPHARM, and Sanofi-Aventis; participating as a member in a company advisory board, board of directors, or other similar group for Actelion, Bayer, Biogen, Clene, Hoffman La-Roche, Merck Serono, MedDay, Novartis, and Sanofi-Aventis; and serving on company-sponsored speaker’s bureau for Sanofi-Genzyme. M.G. reports consulting fees from Biogen, EMD Serono, Novartis, and Sanofi-Genzyme; research support from Alkermes; and speakers’ bureaus for Biogen, EMD Serono, Novartis, Sanofi-Genzyme, and Teva. T.Z. reports fees for participation in scientific advisory boards from Bayer, Biogen Idec, Merck Serono, Novartis, Teva, Genzyme, and Synthon; speaker honorarium from Almirall, Bayer, Biogen, Genzyme, GlaxoSmithKline, Merck Sharp & Dohme, Novartis, Sanofi, and Teva; and research support from Bayer, Biogen, Genzyme, Novartis, Sanofi, and Teva. B.K., I.B., M.L.-B., N.N., D.R., and R.L.-P. report being full-time employees of and holding stock/stock options in Alkermes Inc. L.Y., H.C., S.L., J.H., and C.M. report being a full-time employees of and holding stock/stock options in Biogen.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was funded by Alkermes Inc (Waltham, MA, USA) and Biogen (Cambridge, MA, USA).
Data sharing statement
EVOLVE-MS-1 was registered with ClinicalTrials.gov (NCT02634307). Study data will be shared in accordance with applicable regulations and laws.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.