
Abstract
Background:
Multiple sclerosis (MS) is a complex disease resulting from the joint effect of many genes. It has been speculated that rare variants might explain part of the missing heritability of MS.
Objective:
To identify rare coding genetic variants by analyzing a large MS pedigree with 11 affected individuals in several generations.
Methods:
Genome-wide linkage screen and whole exome sequencing (WES) were performed to identify novel coding variants in the shared region(s) and in the known 110 MS risk loci. The candidate variants were then assessed in 591 MS patients and 3169 controls.
Results:
Suggestive evidence for linkage was obtained to 7q11.22-q11.23. In WES data, a rare missense variant p.R183C in FKBP6 was identified that segregated with the disease in this family. The minor allele frequency was higher in an independent cohort of MS patients than in healthy controls (1.27% vs 0.95%), but not significant (odds ratio (OR) = 1.33 (95% confidence interval (CI): 0.8–2.4), p = 0.31).
Conclusion:
The rare missense variant in FKBP6 was identified in a large Dutch MS family segregating with the disease. This association to MS was not found in an independent MS cohort. Overall, genome-wide studies in larger cohorts are needed to adequately investigate the role of rare variants in MS risk.
Introduction
Multiple sclerosis (MS) is a complex demyelinating disorder of the central nervous system. While the cause of MS is still unknown, there is overwhelming evidence that genetic factors are involved.1,2 Family studies show that one in five individuals with MS have an affected family member, and that the chances of developing MS are 20 times higher for a first-degree relative than for an individual from the general population.3,4
The main genetic locus of MS risk in the North-European population is the human leukocyte antigen (HLA) class II region (the classical HLA-DRB1*15:01 allele). 5 In addition, since the development of genome-wide association studies (GWAS), many non-HLA genetic variants have been identified.6–9 Associated genetic variants both within and outside HLA regions are seen more frequently in familial MS than in MS patients with no family history of MS.10,11
Despite the success in the discovery of new associated loci, the variants in the HLA-class II region and the variants identified by GWAS can to date only explain about 27% of the heritability of MS. 9 More importantly, they do not explain MS in families in which the disease segregates as a major locus with a large effect on the disease risk. It has been speculated that rare variants might explain part of the missing heritability of MS, in particular in atypical familial cases.12,13 Although families with multiple affected individuals are extremely rare in MS, 14 these families may be relevant to investigate and can help in the search to identify new pathways that until now remained undetected in GWAS. Indeed, there are a few reports of whole exome sequencing (WES) successfully identifying rare variants in several MS families.15–19
To identify new rare and coding variants involved in the familial aggregation of MS, we performed WES in a large Dutch MS family with 11 affected individuals. We also investigated whether the variants identified in this family could be found in a cohort of unrelated MS patients and healthy controls.
Materials and methods
Study subjects
A multi-incident MS family was ascertained in 2003 through the still ongoing longitudinal study on Genes and Environment interaction in MS (GEMS) in the Netherlands and has been evaluated for the incidence of new MS cases over the past 13 years. The family originated from the North-East part of the Netherlands, and there was no consanguinity. The number of MS cases was 11 (Figure 1). The MS diagnosis was evaluated according to the standard diagnostic criteria.20,21 The medical ethics committee of Erasmus Medical Center (EMC) approved this study. Written informed consent was obtained from all participants.
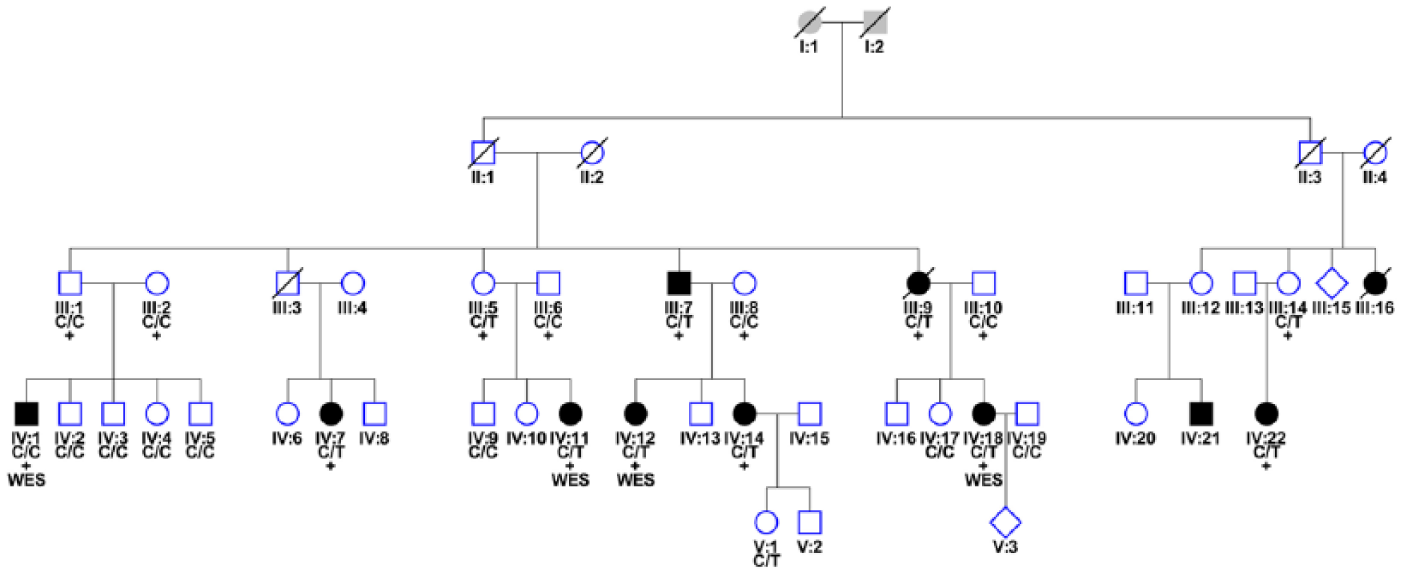
Pedigree of the Dutch family with high prevalence of MS. Filled symbols represent affected individuals. Squares represent males, circles represent females and diamonds represent individuals of an unknown gender. Symbols with a diagonal line represent deceased individuals.
Findings were assessed in a cohort of 591 Dutch patients and 3169 unrelated Dutch controls. The clinical characteristics of the MS patients are described elsewhere. 11 In the patient group, 251 patients were identified as sporadic cases and 340 patients were identified as multiplex cases from 158 MS multiplex families. Healthy controls were enrolled in the longitudinal Rotterdam Study. 22
Genetic analysis
DNA was available from 24 family members and was isolated by a standardized method. 23 A total of 22 individuals were genotyped on IlluminaCytoSNP-12 array and 2 individuals (III:14 and IV:22; Figure 1) on IlluminaCytoSNP-850K array, as they were included at a later stage. The list of overlapping markers from both arrays was used to calculate logarithm of the odds (LOD) scores with Allegro implemented in the easyLINKAGE-Plus. 24 Single nucleotide polymorphisms (SNPs) with a call rate <95% and those showing Mendelian inconsistencies were excluded from the calculations. MS patients were treated as affected, non-carrier family members were considered as healthy, and unaffected mutation carriers with affected children were defined as having an unknown disease status. A multipoint parametric linkage analysis was performed with Allegro with genotypes from 16 individuals (Figure 1) with an SNP spacing of 0.2 and 0.5 cM. LOD scores in sets of 100 markers were calculated assuming the disease in this family to be an autosomal dominant disorder with a population risk allele frequency of 0.001, a penetrance of 70%, and a phenocopy rate of 0.001. 14 Regions showing an LOD score >2.0 were used as candidate regions. Flanking SNP markers were used as borders of the haplotype-sharing regions.
Four distantly related individuals were whole exome sequenced (Figure 1) on the IlluminaHiSeq2000 platform using Agilent’s SureSelect All Exon Kit. Variants with a minor allele frequency (MAF) of ⩽1% were searched (see supplementary data for details). Candidate single nucleotide variants (SNVs) from the WES analysis were then validated in all available family members using iPLEX Gold assays on a MassARRAY system (Agena Bioscience, San Diego, CA, USA). Samples with variant call rate lower than 50% were discarded. The average genotype call rate was 98.9%. Array-based genotypes of independent Dutch MS cases and healthy controls from the Rotterdam study were available for a secondary analysis.
Results
Characteristics of the affected family members
Clinical characteristics of MS patients are presented in Table 1. The MS patients were 47.9 ± 16.3 years old. Their mean age at the disease onset was 31.3 ± 14.0 years, and the male to female ratio was 1:3.5. The median disease duration was 16.6 years (ranged from 1.5 to 28.8 years). The median Expanded Disability Status Scale (EDSS) score was 3.5 (mean 4.7 standard deviation (SD) ± 3.13) and the mean MS Severity Score (MSSS) was 5.3 (SD ±3.26). MS with a bout-onset was predominantly represented (i.e. relapsing remitting MS, n = 5; secondary progressive MS, n = 3; two patients had an unknown disease course and one patient had primary progressive MS). We interviewed seemingly healthy individuals by phone for disease symptoms, none of these showed evidence for MS based on signs and symptoms.
Clinical characteristics of MS patients from the MS family.
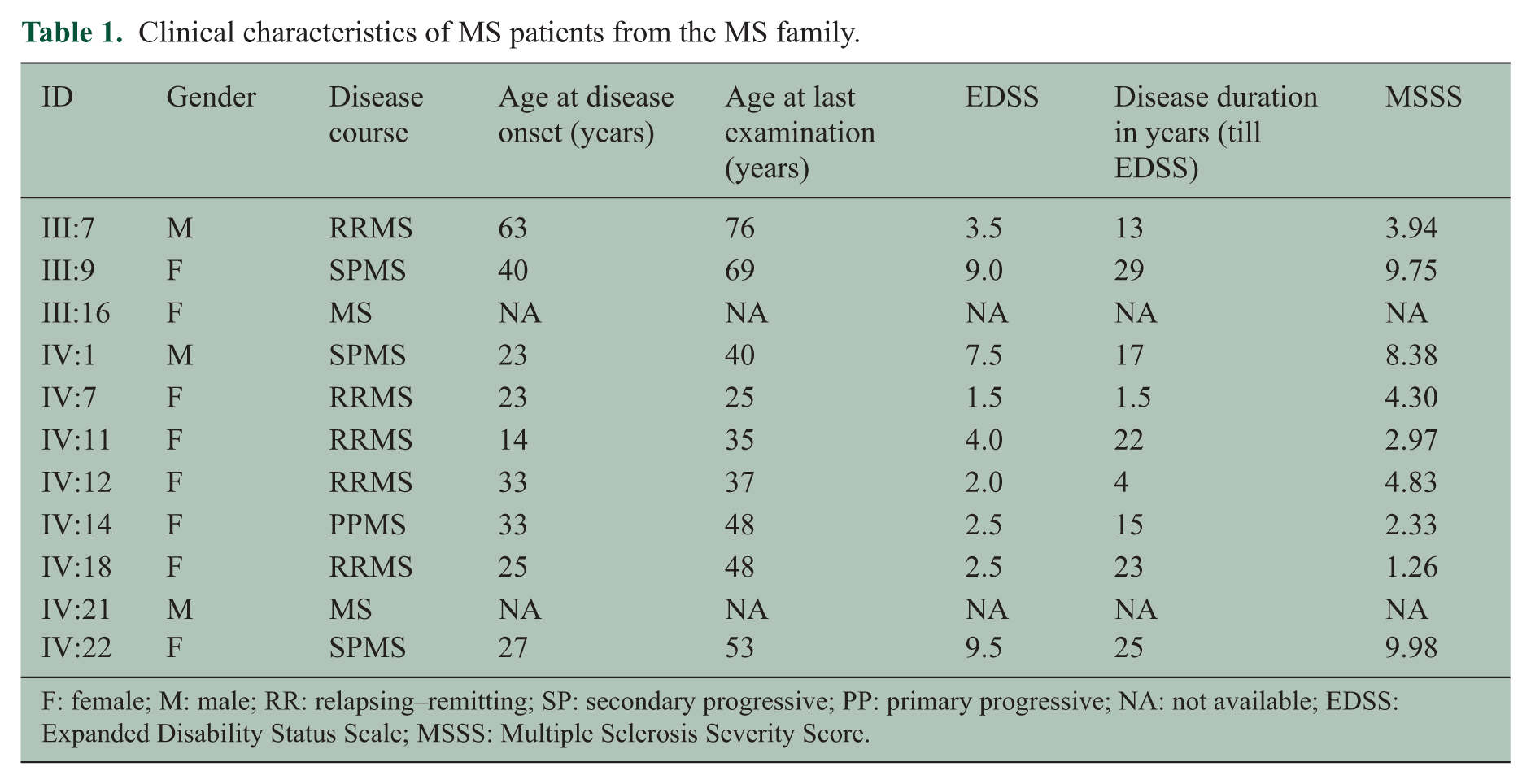
F: female; M: male; RR: relapsing–remitting; SP: secondary progressive; PP: primary progressive; NA: not available; EDSS: Expanded Disability Status Scale; MSSS: Multiple Sclerosis Severity Score.
Genetic analysis
Multi-point parametric linkage analysis of 16 patients (of which 9 with MS) resulted in a maximum LOD score of 3.0 on chromosome 7 (Supplementary Figure 1). Analysis of chromosome 7 revealed a shared haplotype region between flanking SNPs rs11972782 and rs6959538 (Human GRCh 37/hg19, chr7:69877261-73139762) in all but one affected individual (IV:1, Figure 1). Four distantly related individuals were then whole exome sequenced. WES generated 5.1 Gb of reads per individual. The mean coverage was 44×, and at least 20× of coverage was achieved for 71% of the exome for four samples. On average, 35,313 variants (range = 33,840–36,087) were called per individual after quality control. To reduce the number of variants, we filtered the WES data by narrowing the genomic region to the haplotype shared region found in the linkage analysis and setting the MAF at ⩽1% in 1000 Genomes Project and Exome Sequencing Project (ESP) database. Further analysis of exonic and splice variants containing non-synonymous and stop variants revealed only one rare non-synonymous SNV rs147213094 (C/T, located on chr7:72745738MAF in ESP = 0.0041, gnomAD = 0.002962, ExAC = 0.003155) in the FKBP6 gene, shared by three of the four sequenced individuals. Other chromosomes were analyzed in WES with the above-mentioned criteria, and MAF between 1% and 5% was also used. Several potential candidate variants in the following genes shared by three and four sequenced individuals were found: FAM184B (rs61741403), ACOT12 (rs34607174), PPP1R9A (rs61737465), ADAMTS13 (rs28503257), SPTY2D1 (rs35411689), ZNF750 (rs35283702), MED13L (rs113890513), and ABCC3 (rs11568591). By means of sequenom analysis, the variants were assessed in four WES sequenced individuals and genotyped in the remaining family members. Only the SNP rs147213094 in the FKBP6 gene located in the linkage region segregated with MS in this family and was present in 8/9 (88.9%; one affected individual (IV:1) that did not share the haplotype also did not carry the FKBP6 risk variant) of the affected individuals (with DNA available). There were 3/15 (20%) carriers of this variant among unaffected individuals (with DNA available). A healthy female (V:1) carried the risk allele and was according to her age still at risk for MS. Two other unaffected individuals were parents and obligate carriers (III:5 and III:14, respectively) who transmitted the allele to their affected offspring.
There were no differences in the HLA-class II risk allele frequency between cases and unaffected family members (p = 0.23, Table 2). After correcting for the HLA-DRB1 status, MS patients carried significantly more often the T-allele than the healthy individuals in this family (odds ratio (OR) = 6.9 (95% confidence interval (CI) = 1.2–39.8, p = 0.03). Furthermore, MS patients with the T-allele (minor allele of FKBP6) did not differ in their HLA-DRB1 status compared to MS patients that did not carry the FKBP6 minor allele (p = 0.06).
Genotypes of MS patients and their family members.
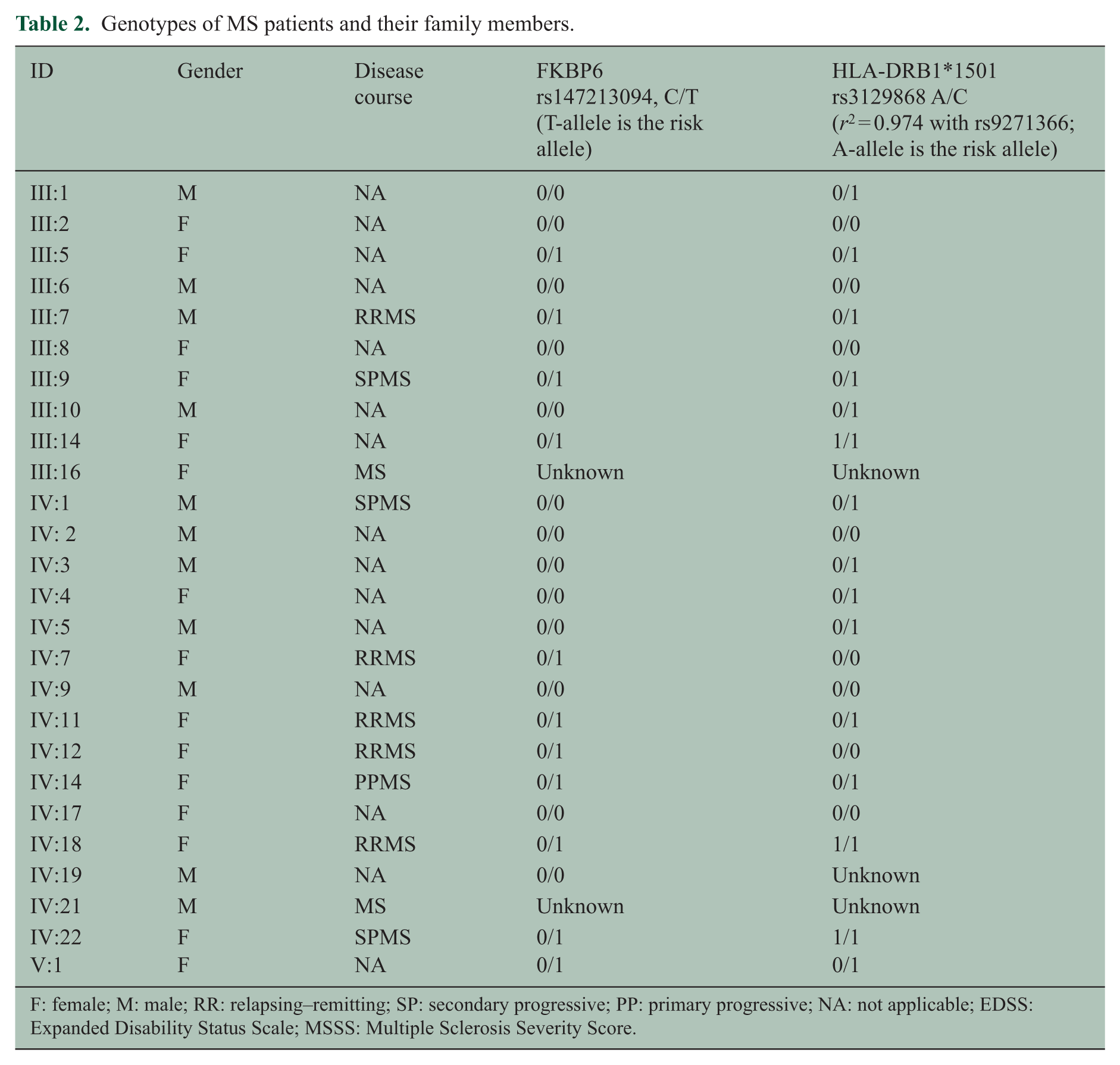
F: female; M: male; RR: relapsing–remitting; SP: secondary progressive; PP: primary progressive; NA: not applicable; EDSS: Expanded Disability Status Scale; MSSS: Multiple Sclerosis Severity Score.
The frequency of rs147213094 was further analyzed in 591 MS cases and 3169 healthy controls. The MAF was slightly higher in all MS patients than in healthy controls (15/1182 alleles (MAF = 1.27%) vs 60/6338 alleles (MAF = 0.95%), respectively), but there was no significant association with the disease risk (OR = 1.33 (95% CI = 0.8–2.4), p = 0.31). In the patient group, 251 patients were identified as sporadic cases and 340 patients were identified as multiplex cases from 158 MS multiplex families. Sporadic MS patients had MAF of 0.8% (4/502 alleles) and multiplex patients had MAF of 1.62% (11/680 alleles) of rs147213094. Taking only the probands from the MS multiplex families (n = 158, 8/316 with MAF = 2.5%) revealed a marginal significant association with the disease risk (OR = 2.7 (95% CI = 1.3–5.7), p = 0.01 with a MAF in patients of 2.5% compared to 0.95% in general Dutch population). This is, however, a a-posteriori analysis that lacks genome-wide significance and adjustments for sex, population structure, and MS polygenic risk score based on approximately 200 common susceptibility loci in and outside the HLA-class II region. The variant was enriched in Dutch controls compared to the low frequency in gnomAD (MAF = 0.002962)and ExAc database (MAF = 0.003155).
Frameshift insertions, deletions, splicesite mutations, stopgain, and stoploss variants located in the linkage region and genome-wide shared by sequenced individuals were inspected, but no potential candidates were identified.
There were no rare damaging variants in the 110 MS risk genes identified by the recent MS GWAS 9 shared by sequenced individuals.
In earlier few WES studies in MS families, several rare variants were reported: a p.A53T variant in TYK2 (rs55762744), 15 a p.R389H variant in CYP27B1 (rs118204009),16,17 a p.G420D variant in PLG (rs139071351), 19 and a p.Arg415G in variant in NR1 H3(rs61731956). 18 All variants were found in the Canadian MS patients. None could be found in our WES data.
The SNV rs147213094 (C/T) encodes a missense mutation in exon 5 of FKBP6 that changes arginine into cysteine (p.R183C). The variant overlaps several transcripts and in many instances tagged as a damaging variant by multiple algorithms including PolyPhen, SIFT, MetalLR, MetaSVM, Mutation Taster, and PROVEAN, but not by InterVar that classifies this variant as “benign” (according to 2015 American College of Medical Genetics Criteria). The computational tools for measurement of evolutionary constraint showed an elevated level of conservation for the arginine residue in mammals (Genomic Evolutionary Rate Profiling (GERP)++ = 5.46; PhyloP = 2.35; SiPhy = 8.64), indicating that this amino acid is important for protein function (Supplementary Figure 2). Additional support is provided by the Combined Annotation Dependent Depletion (CADD) score (19.6) indicating that the amino acid substitution belongs to the 10% of the most deleterious substitutions genome wide.
The messenger RNA (mRNA) of FKBP6 gene is broadly expressed in all tissues, being the highest in the testes (Figure 2).
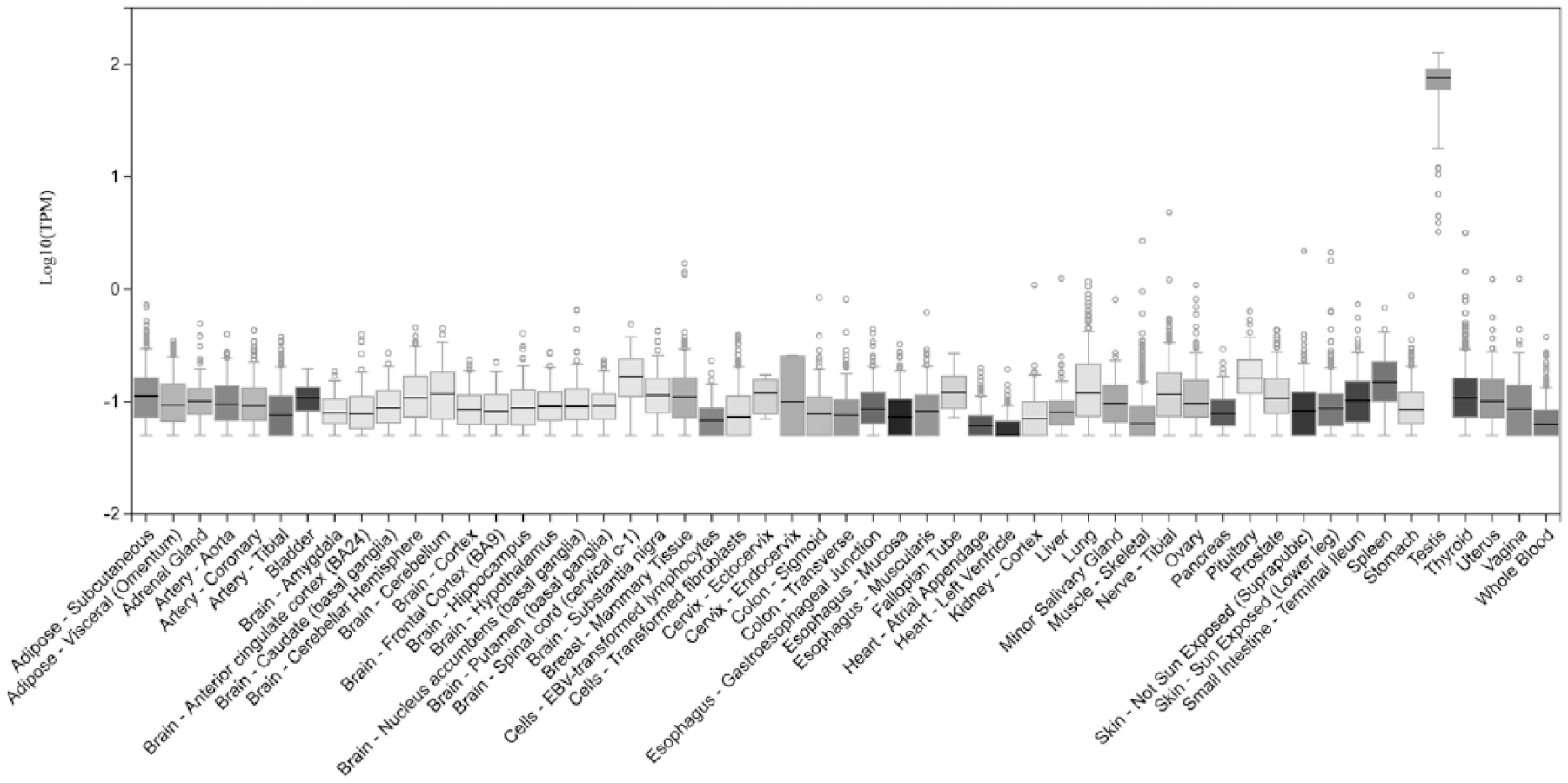
Expression of the FKBP6 from the GTEX database with highest expression in the testes.
Analysis of protein–protein interactions by STRING (software version 10.0) highlighted the interaction of FKBP6 with glyceraldehyde 3-phosphate dehydrogenase (GAPDH) and heat shock protein (HSP) 90 (Figure 3). 25 The included pathways are regulation of nitric oxide biosynthesis, estrogen signaling pathway, and major histocompatibility complex (MHC)-class II protein binding.
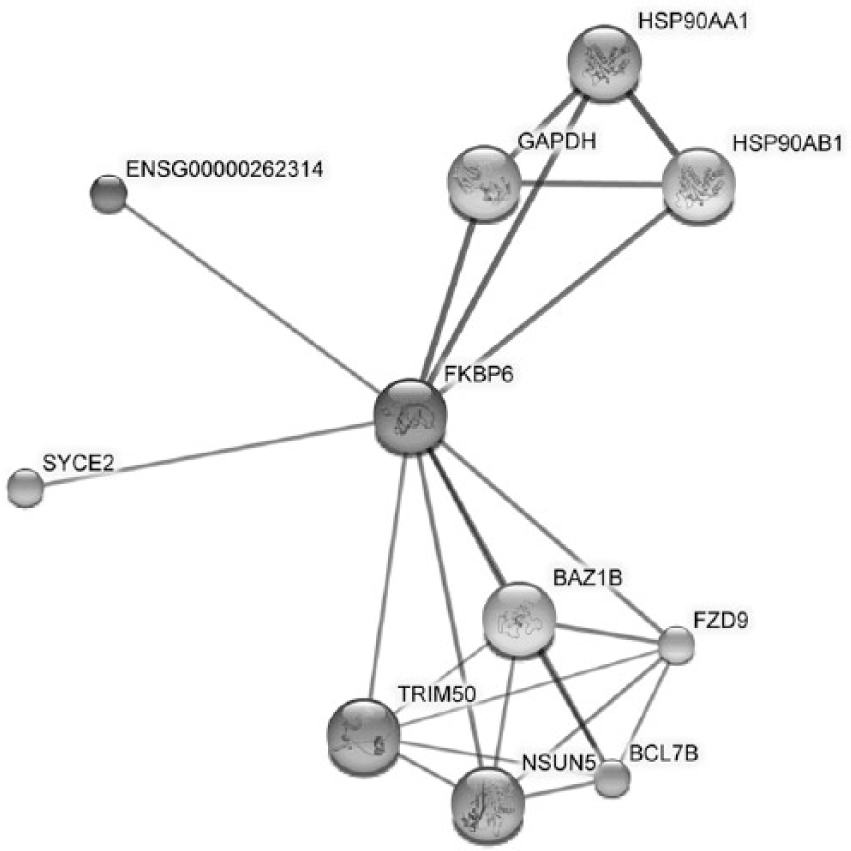
Analysis performed by STRING software (version 10.0). Thicker lines indicate more confident protein–protein interactions.
Discussion
We found a rare missense variant (rs147213094) in the FKBP6 gene in a large Dutch MS family which resides in the linkage area in the chromosomal region 7q11.22–q11.23. We identified this variant following a classical linkage analysis that revealed a suggestive evidence for linkage to the 7q11 region. This variant was shared by three of the four relatives who were exome sequenced. In the same WES data searching for variants shared by three and four sequenced individuals other eight variants emerged; however, only FKBP6 was found to segregate with MS when extending the family to nine cases (eight of nine were carriers). One individual with MS did not carry the risk variant. Probably other, non-genetic factors are involved in this individual, because MS has a multi-factorial nature. While the variant showed good segregation pattern in the family, its association outside the family was not significant in a moderate-sized validation cohort of cases and controls. In a secondary analysis, an increased frequency of the variant was found in the probands and in the total multiplex group, but not in sporadic patients. This observation, however, lacks genome-wide significance in this moderate-sized population and statistical adjustments have not been possible for sex, population structure, and MS polygenic risk score. Despite the marginal enrichment of this variant in the multiplex MS, the hypothesis that this rare variant in FKBP6 gene is more representative for multiplex MS and not for sporadic MS per se in Dutch population cannot be claimed here. Validation in other and larger cohorts of multiplex MS families will be needed.
The FKBP6 gene is located on chromosome 7 and belongs to the immunophilins FK-506 binding protein (FKBP) family of highly conserved proteins which possess binding abilities to immunosuppressive drugs. FKBP6 was first described in the context of Williams–Beuren syndrome (WBS), a genomic disorder with congenital cardiovascular defects, dysmorphic facial features, mental retardation, azoospermie, and hypercalcemia caused by a hemizygous contiguous gene deletion on chromosome 7q11.23 encompassing 28 genes in the largest deletions, including FKBP6. 26 But also smaller deletions are found in patients excluding FKBP6. 27 FKBP6 was identified as a component of the synaptonemal complex that forms between two homologous chromosomes during meiosis and mediates chromosome pairing, synapsis, and genetic recombination. Four missense variants in FKBP6 have been associated with human male infertility in a study in idiopathic infertile men. 28 A recent paper identified that the co-chaperone FKBP6 acts as a host factor required for hepatitis C virus (HCV) replication, that is, HCV replication was completely suppressed in FKBP6-knockout hepatoma cell lines, while the expression of FKBP6 restored HCV replication in FKBP6-knockout cells. 29 However, we speculate that FKBP6 might have as yet undiscovered functions.
The FKBP6 gene is not among the known MS risk loci. There is some evidence for its possible role in MS susceptibility. The chromosomal region 7q11–q21 has previously displayed a suggestive linkage to MS with a LOD = 1.14 in 52 multiplex families (31% with multiple affected generations and/or affected collateral relatives) of European descent. 30 In the experimental autoimmune encephalomyelitis (EAE) model in rats, there was a significant linkage in the chromosomal region analogous to the human region containing FKBP6 with the incidence and duration of EAE and also the maximum EAE score. 31 A case report described a co-occurrence of WBS and MS in an adolescent patient. 32
To further explore how FKBP6 might be linked to MS, we studied pathways and interactions of FKPB6 protein with other proteins. Analysis of protein–protein interactions by STRING highlighted the interaction of FKBP6 with GAPDH and Hsp90. Studies have shown that FKBP6 (also known as FKBP36) can inhibit GAPDH activity and expression. 33 When GAPDH enzyme activity is inhibited, neurons display chromatin condensation, internucleosomal DNA cleavage, and cytoplasmic shrinking, 34 and this could result in widespread neuroaxonal apoptosis and degeneration. FKBP6 also interacts with Hsp90 by being its co-chaperone, and this complex can interact with different proteins.
The rs147213094 in FKBP6 encodes a missense mutation that changes arginine to cysteine (R183C). The variant is located in exon 5 and expected to affect the tetratricopeptide (TPR) repeat of the FKBP6 protein. The TPR repeat domains are critical for transmembrane protein–protein interactions and the formation of multiprotein complexes. 35 It is as yet not clear how the identified polymorphism might affect the protein–protein interaction process and be involved in MS susceptibility.
We failed to identify new rare variants in the 110 MS GWAS loci and previously reported rare variants in TYK2, CYP27B1, PLG, and NR1H3 in the exome-sequenced patients.15,16,18,19 Concerning the rare p.R389H mutation in CYP27B1 found in Canadian families, 16 only one study could replicate this variant in a multi-incident MS family of the Canadian descent, but not in the general Canadian MS population. 17 Other studies failed to replicate this finding in ethnically different populations.36,37 There are no studies we are aware of that tried to replicate the rare variants in TYK2 15 and PLG. 19 A recent publication of a rare variant in NR1H3, which claimed that this variant was causal for familial MS and a common variant in the same gene was associated with primary progressive MS, caused a discussion in the scientific community38,39 with several parties debating the arguments pro and against its role in MS. It is very likely that the rare variants are family- and population specific and have therefore different distributions in different populations compared to common variants. 40 Rare variants can also show higher levels of stratification and therefore lack replication in association studies. 40 Systematic interrogation of genome-wide rare variants in larger cohorts is needed to address this issue properly.
There were several limitations to our study. First, by finding a suggestive linkage on chromosome 7 and combining these data with WES, we were able to point to an SNV with medium increased risk that turned out to be segregating with the disease in this family. The search for rare variants in other regions and the MS GWAS loci relied on the analysis of only four exome-sequenced individuals. If there were more rare SNVs with medium increasing MS risk, these would probably be missed because of only few individuals WES analyzed. Second, because we are dealing with a rare variant, the number of patients in the validation cohort should be preferably in the thousands. The major limitation of this study is lack of power in the validation cohort due to limited patient resources.
In summary, we here report a rare variant in the FKBP6 gene found in a Dutch multi-incident MS family using linkage analysis and whole exome sequencing technology. Evidence for its involvement in familial MS in general is not sufficiently supported here. The few studies thus far that claimed the influence of rare genetic variants on MS susceptibility are still devoid of replication.36–39 Systematic interrogation of genome-wide rare variants in larger cohorts is needed to answer the question if and to what extent rare variants contribute to MS risk.
Supplemental Material
MSJ777202_supplementary_data – Supplemental material for Linkage analysis and whole exome sequencing identify a novel candidate gene in a Dutch multiple sclerosis family
Supplemental material, MSJ777202_supplementary_data for Linkage analysis and whole exome sequencing identify a novel candidate gene in a Dutch multiple sclerosis family by Julia Y Mescheriakova, Annemieke JMH Verkerk, Najaf Amin, André G Uitterlinden, Cornelia M van Duijn and Rogier Q Hintzen in Multiple Sclerosis Journal
Footnotes
Acknowledgements
The authors would like to thank all members of the family, other MS patients, and healthy controls who participated in the study. Authors would also like to thank MMPJ Verbiest for sample handling and technical assistance.
Declaration of Conflicting Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: R.Q.H. has participated in trials with Biogen Idec, Merck Serono, Roche, and Novartis and serves on the editorial board of Multiple Sclerosis and Related Disorders. The remaining authors report no conflict of interest.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The MS center ErasMS is financially supported by the MS Research Foundation of the Netherlands.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.