
Abstract
This work presents an enhanced identification procedure utilising bioinformatics data, employing optimisation techniques to tackle crucial difficulties in healthcare operations. A system model is designed to tackle essential difficulties by analysing major contributions, including risk factors, data integration and interpretation, error rates and data wastage and gain. Furthermore, all essential aspects are integrated with deep learning optimisation, encompassing data normalisation and hybrid learning methodologies to efficiently manage large-scale data, resulting in personalised healthcare solutions. The implementation of the suggested technology in real time addresses the significant disparity between data-driven and healthcare applications, hence facilitating the seamless integration of genetic insights. The contributions are illustrated in real time, and the results are presented through simulation experiments encompassing 4 scenarios and 2 case studies. Consequently, the comparison research reveals that the efficacy of bioinformatics for enhancing routes stands at 7%, while complexity diminish to 1%, thereby indicating that healthcare operations can be transformed by computational biology.
Introduction
Given that contemporary healthcare institutions prioritise data-driven computer vision methodologies, it is essential to develop a cohesive strategy that incorporates computational techniques for effective prediction and classification. In contemporary healthcare, data-driven methodologies can be developed through bioinformatics, which entails the discovery of genes utilising biological data. The utilisation of biological data not only clarifies the identification process but also significantly contributes to elucidating complex mechanisms that can regulate disease propagation factors. Furthermore, bioinformatics is recognised as an efficient data management technique for handling substantial volumes of data associated with biological parameters (genes) and for next-generation healthcare networks that necessitate genetic markers. The periodic identification of diverse genes has been enhanced for bioinformatics, therefore reducing off-target effects, so boosting safety and facilitating effective gene tracking. Consequently, the expedited method of gene identification through bioinformatics facilitates vaccine production and reveals concealed datasets by utilising medical imaging, thereby mitigating danger factors for individuals. In addition to gene identification, the integration of bioinformatics with various optimisation algorithms is crucial for ensuring standardisation and data privacy; thus, the most effective deep learning optimisations can be combined to enhance capabilities in healthcare identification. Nonetheless, a primary restriction of bioinformatics is the necessity to provide scalable infrastructures for effective gene identification, hence necessitating multi-dimensional approaches in healthcare.
Figure 1 depicts the block diagram of bioinformatics in healthcare, integrated with deep learning optimisation. Figure 1 illustrates that all essential components, including data sources, data pre-processing units and bioinformatics tools, are aggregated and linked to acquire clinical data from other interconnected units to commence gene identification. During data preparation, genes are normalised, and other relevant properties are retrieved for subsequent sequencing using bioinformatics techniques, hence achieving regularised data. In contrast, the regularised genomic data undergoes additional processing for acquisition and collection; for large-scale data, it is essential to implement scalable computer resources by establishing a separate infrastructure unit. Consequently, the accessible genetic data will be modelled by deep learning optimisation, which will subsequently be validated and processed for informed decision-making.

Block diagram of bioinformatics with optimisation in health care.
Background and related works
This section provides a thorough analysis of bioinformatics in healthcare to elucidate the issue areas addressed by current methodologies. To implement the suggested task in real time, it is essential to establish a solid foundation in the fundamental principles of gene identification. The identification process that facilitates prediction, categorisation and treatment is essential, and addressing the limits of current methodologies is recognised for developing the system model and analysing the associated outcomes. In Ref., 1 a digital filtering and decomposition method that enables adaptive processing within body area networks is employed for healthcare applications to isolate data signals utilised for gene identification. The gathered genetic data effectively mitigates the impact of unwanted alterations, resulting in a significant reduction in variances in gene identification and thereby diminishing computational difficulties. Although undesirable modifications are minimised in this instance, it is essential to breakdown the numerous signals available in individual states, so creating new spaces distinct from the original genetic data representations. The historical advancements of developing trends in biomedical research are contrasted, and the advances implemented for personalised medicine significantly advance main computational approaches. 2 Conversely, gene identification enhances patient care by facilitating early diagnoses; thus, it is essential to integrate data to achieve high-quality omnic representations, enabling more trustworthy interpretations at this stage. However, a co-relationship metre must be established for each process, as meta-analytic outcomes can only be achieved by augmenting the interference in each genetic dataset, thus resulting in an increase in error circumstances.
A knowledge-based framework is established in Ref. 3 for analysing gene features by incorporating compression techniques, with selected parameters assessed according to specific criteria. Characteristic analysis addresses all biological issues by integrating input from various users, leading to genetic identifications derived from an inferential approach. Nonetheless, only undesirable answers are obtained in these instances, as the classification of the knowledge-based system varies with the parameters studied; thus, it is essential to employ only generalisation approaches for subsequent research. In addition to characteristic analysis, the application of artificial intelligence algorithms for gene analysis is demonstrated through the examination of 2 public datasets, establishing linear correlations in this context. 4 The linear linkages among data sets enhance the potential for uncovering novel solutions, leading to global resolutions for diverse phenotypes. The primary challenge in the discovery process is that each metabolic profile significantly influences outcomes, making standardisation across diverse genetic factors exceedingly challenging. Enhanced forecasts are then generated by machine learning approaches to improve individual health situations, since early case predictions are established by analysing each gene. 5 This style of prediction employs strongly additive explanations to produce a trade-off between genetic insights and transparency, hence enhancing interpretability. Moreover, actionable insights indicate that alterations or combinations of genes may differ, resulting in significant impacts due to the identification of extraneous data.
Consequently, all open statement concerns with gene identification in healthcare applications are analysed, enabling the provision of effective management solutions even in the face of extensive information. 6 Increased data volume enables the development of extensive architectures that connect data with potential interdependencies, facilitating user sharing of available genetic information. In the presence of accessible open data, all genetic information will be expunged from the underlying data management, resulting in alterations to the original gene information; consequently, only singular point entries will be delineated. Furthermore, novel sequencing technologies will provide the storage of greater volumes of genetic information to enhance healthcare quality. 7 It is noted that the extensive number of discoveries renders the assumption regarding the adoption of appropriate approaches irrelevant concerning various gene sequences. Similarly, even when sophisticated procedures are employed for loading gene sequences, the overall mistake rate escalates, resulting in novel gene patterns that cannot be processed at intermediate stages. Conversely, the future prospects for healthcare that analyse each gene’s information through the Internet of Things can yield significant data by employing appropriate architectures. 8 The analysis of data at this stage necessitates advanced computational methods, which can be adequately supported by suitable infrastructure and relevant genetic elements. In the aforementioned scenario, extensions may be facilitated through the implementation of classification algorithms, hence enabling case study control to delineate each gene; such controls are necessary alone when variable selection methods are employed. 9 Moreover, only case-control studies are conducted for each gene using accurate labels, and the identification procedure stays consistent with other scenarios. Table 1 presents a comparison of the existing and suggested methods by evaluating several criteria.
Existing versus proposed.
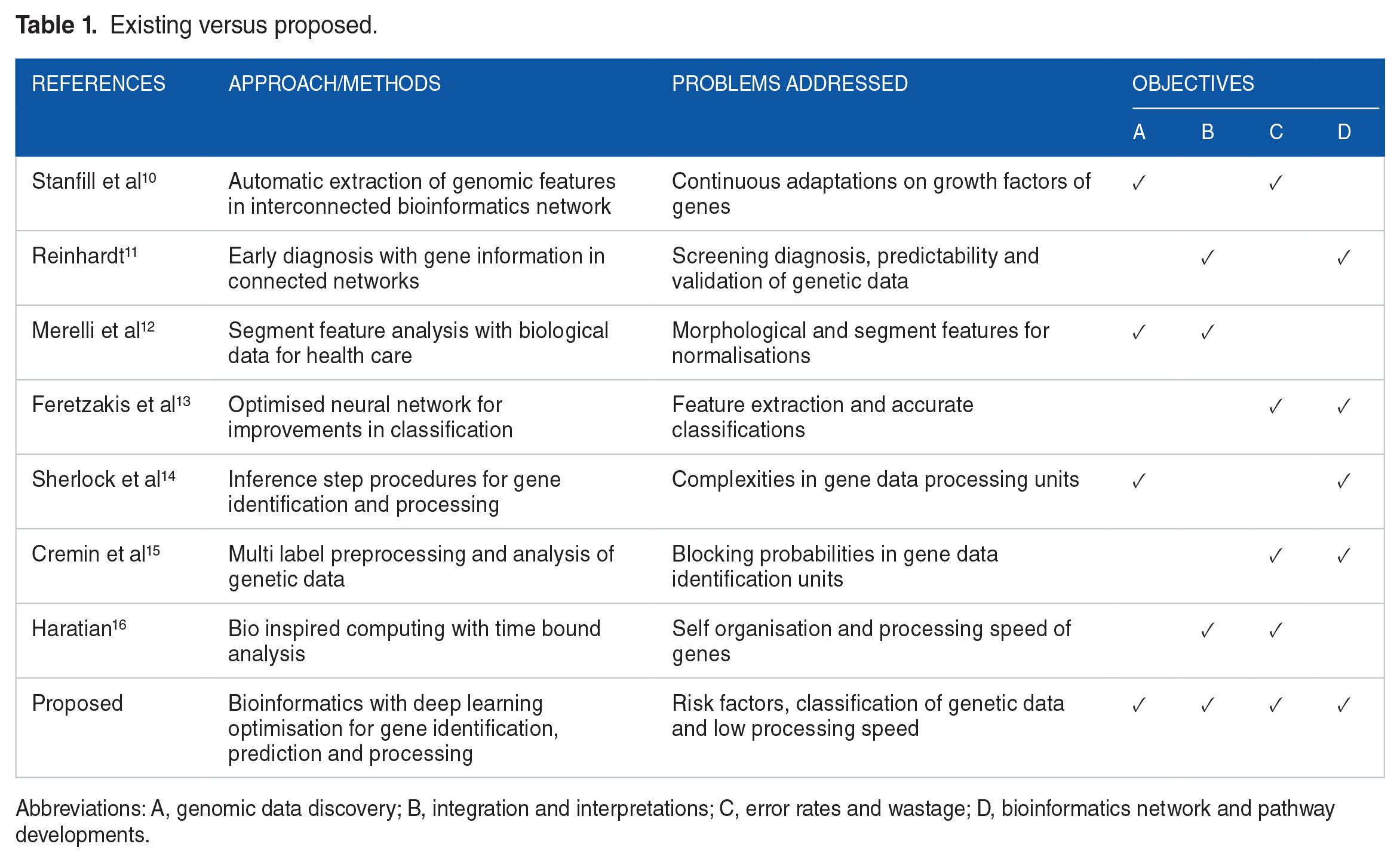
Abbreviations: A, genomic data discovery; B, integration and interpretations; C, error rates and wastage; D, bioinformatics network and pathway developments.
Research gap and motivation
Despite the introduction of numerous strategies for gene identification in health care, as outlined in Table 1, the integration of varied data from various sources remains a very difficult function, consequently exacerbating gaps in existing health care operations. The unresolved gap prolongs the processes of prediction, categorisation and other activities necessary for detecting various disorders, hence adversely impacting individual health. Furthermore, the real-time implementation of current methodologies is constrained in terms of generalisation, necessitating substantial CPU resources for processing. In addition to computing resources, the challenges of integration and interpretation persist, even with the introduction of decision support systems, since increased errors and data wastage are noted. Consequently, based on specific observations, the gap in current studies concerning personalised healthcare and bioinformatics raises 3 enquiries as follows.
RG1: Can genomic data be introduced with minimal risk factors, normalisations and error rates?
RG2: Is it possible to integrate and understand the data effectively while minimising data waste management?
RG3: Does deep learning optimisation offer an effective method for constructing the bioinformatics network with maximised benefits?
Major contributions
The suggested method emphasises the integration of deep learning optimisation that incorporates both normalisation and hybrid techniques to enhance the potential for gene mapping through subsequent parametric analysis.
To establish a framework for the integration of multi-omics data by incorporating genomic data with reduced risk and minimised error rates.
To minimise data wastage by identifying appropriate physical representations for real-time notifications.
To establish a secure bioinformatics network that optimises benefits through the advancement of individual pathways.
Proposed System Model
This section discusses the analytical representations of medical healthcare related to bioinformatics, aimed at transforming data to exploit diverse scenarios for achieving real-time outcomes. The suggested system model supplies essential information for data integration and interpretation, enabling the identification of genetic variables through a systematic approach. Conversely, with an appropriately designed system model featuring relevant analytical equations, the complexity of prediction and risk assessments can be decreased.
Genomic data
To integrate genetic data into healthcare operations, it is essential to mitigate the hazards associated with processing units. Consequently, each individual can analyse real-time representations by delineating numerous healthcare characteristics and associated coefficients with hereditary components, as specified in equation (1).

Where,
Equation (1) establishes that the parameters related to health care will significantly impact the formation of similar probability conditions. Furthermore, the relevant data factors in each instance are utilised to mitigate risk factors, hence facilitating successful data processing.
Preliminary 1
Let us examine the comprehensive risk variables linked with each health monitoring metric as

Lemma 1
To establish the correlation between risk factors and genetic data, it is imperative to address linearity issues, and each healthcare concern must be delineated according to the principle of linear combinations. Thus, linear representations enable the simplification of the decision variables as illustrated in equation (3).

Data integration and interpretation
To optimise parameters effectively in health care operations, it is crucial to integrate diverse data sources and analyse the numerous hazards associated with each gene. Therefore, all physical activities must be tracked with the relevant external elements as delineated in equation (4).

Where,
Equation (4) indicates that the aggregated data can be collected and represented using large data units, therefore enhancing health outcomes. Consequently, at a later stage, each user can reference the current state of representations in relation to earlier units, thereby mitigating risk factors during the pre-processing phase.
Preliminary 2
Let us examine the normalisation and standardised representation of diverse data across numerous scales as

Lemma 2
To validate the data integration and interpretation methods, multivariate principal analysis must be employed, thereby accurately delineating the statistical representations of each variable. Furthermore, homogeneous data must adhere to the interaction processes specified in equation (6).

Error rates
Error representations in each healthcare unit are identified by determining potential error rates, hence preventing the loss of biological data. To analyse diverse parameters specified by many data sets, it is essential to minimise error, and the model for such representations is articulated in equation (7).

Where,
Equation (7) indicates that misclassification mistakes in protected health care data pertaining to genetic information must be minimised. Furthermore, this categorisation method can be streamlined by validating many gene factors organised in a sequential manner.
Preliminary 3
Let us denote the data segments for each gene sequence as

Lemma 3
To validate the error states in alignment with gene indications, it is essential to examine many data points, such as

Data wastage
In healthcare operations, the data pertaining to numerous genetic factors may vary, exhibiting either higher or lower values depending on individual gene variations. Thus, the expenses related to representations and associated wastage must be delineated in accordance with equation (10).
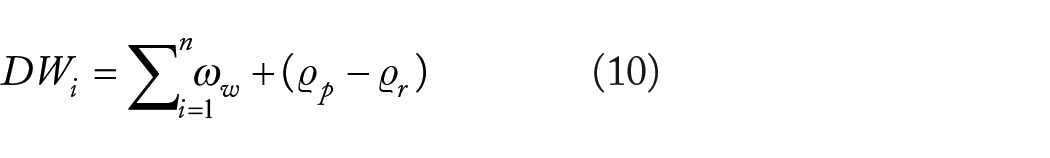
Where,
Equation (10) suggests that if the disparity between prescribed and required data is significantly greater, the penalty factor must be modified. Consequently, to mitigate fines, the genetic data must conform to mandated and specified circumstances, so enhancing overall cost-effectiveness and efficiency.
Preliminary 4
Although genetic health data can facilitate optimisation, it is essential to define specific restrictions by identifying the adjustment parameters. Let us assume the total number of changes as

Lemma 4
To achieve minimal changes for diverse data measurements, it is essential to adhere to genetic test cases by proposing trial-and-error genetic pathways. Consequently, to sustain minimal modifications in unfavourable reactive situations, the minimum and maximum values are specified in equation (12).

Bioinformatics network
As the number of genes seen in a connection network increases, it is essential to facilitate interactions among diverse units to enable recovery. Consequently, in addition to the sequence distribution of genes, an individual gene network that illustrates the similarities among individuals is identified and expressed using equation (13) as follows.

Where,
Equation (13) suggests that enhancing gene similarity can minimise alterations in network decisions, hence optimising the efficiency of the bioinformatics network. Furthermore, the probability of prediction inside each network can be enhanced in this scenario to facilitate interaction among interconnected networks.
Bioinformatics gain
To uncover potential biological characteristics within the examined genetic components, it is essential to enhance overall gain while dealing with complicated data sets. Consequently, specific biomarkers are identified and classification is executed, enhancing the predictive process as demonstrated in equation (14).
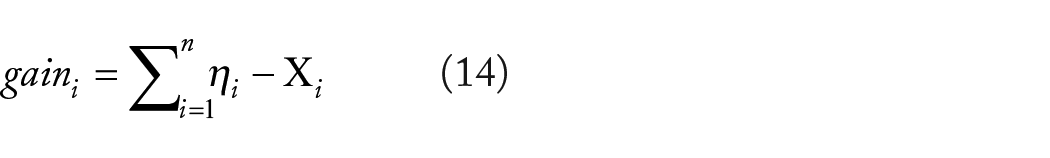
Where,
Equation (14) indicates that if the discrepancies between the total and the separation of data sets are minimal, enhanced predictions are achievable, hence increasing the accuracy of classifications. This method of prediction may eliminate unnecessary features across diverse network distributions, hence attaining a 100% improvement.
Gene pathway development
The biological pathways for forecasting a specific disease are analysed by establishing suitable connections among the interrelated networks. A primary requirement for pathway development is the execution of coordinated actions involving numerous genes, hence facilitating appropriate treatment strategies. Therefore, the score factors for each pathway can be established using equation (15) as follows.

Where,
Equation (15) indicates that the individual weight of genes may solely be ascertained by sequential classification methods, necessitating robust associations for all functional units. Nonetheless, the aforementioned advancements can only be achieved if specific hypotheses are formulated using bioinformatics patterns.
Objective functions
Complex system patterns necessitate the utilisation of a composite objective function to integrate many aspects, hence facilitating balanced healthcare procedures. Thus, the genetic parameters are delineated using min-max criteria, with interpretations simplified as outlined in equation (16).
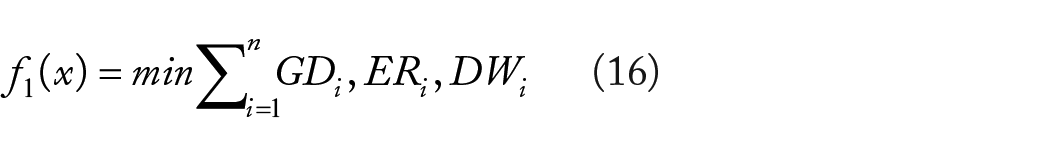

The above mentioned composite objective functions are balanced with separate biological contents where total objective function can be represented as follows.

To enhance the effectiveness of the suggested system model, deep learning optimisation is employed to extract features from image sets for accurate genomic analysis. The delineation of the integrated system model and optimisation method is as follows.
Deep Learning Optimization
Deep learning optimisation is combined to attain comprehensive equilibrium among the evaluated parameters, including error reduction, enhancements in gain and other pertinent criteria. Deep learning optimisation offers distinct weighting elements for modifying intricate issues in healthcare operations, facilitating trade-offs within this integration. The primary benefit of selecting deep learning optimisation for the integration process is the enhancement of prediction and classification capabilities, especially with high-dimensional bioinformatics datasets. Furthermore, deep learning optimisation can identify complicated patterns associated with genetic components, thereby establishing non-linear correlations in a brief timeframe. Multi-modal data can be detected without human feature extraction, enabling comprehensive detection and prevention through deep learning optimisation in healthcare operations. Significant genes associated with distinct diseases are found through the use of relevant image aspects, hence enhancing the generalisation factors in the processing of individual treatments.17,18 The suggested method integrates deep learning models using an 8-step procedure, wherein data is generated and pre-processed at the first stage of operation. Upon data generation, a sequential model will be constructed and compiled according to predefined objective functions, facilitating early detection in the subsequent phase, while appropriate training will be administered for error factors. The subsequent operation entails the testing, assessment, optimisation and comparison of genes; should there be an increase in loss functions, the parameters of each model will be altered to identify the ideal solution. The various types of deep learning optimisation essential for efficient operations are outlined below.
Regularisation technique
In the optimisation process of deep learning, regularisation metrics are crucial as they impose a penalty factor, whereby random units are eliminated from biological data, since genetic variables are often insufficient in attaining the desired results. Consequently, training can be conducted just on specified gene data, and all forms of noise present will be eliminated throughout this process. Additionally, for extensive biological databases, a penalty function can be included to avert the loss of complete gene data; hence, once training concludes, automatic testing activities can be executed. The primary benefit of the regularisation technique is that it enhances generalisation for novel and concealed genetic data by improving feature insights, hence facilitating the automatic selection of key genes. By obtaining generalised data in this instance, a more straightforward solution with automated balance may be offered, thereby entirely eliminating dependencies associated with this regularisation process. Consequently, the regularisation metrics not only integrate a penalty function for the analysed genomic data but also yield credible predictions, enabling precise classifications and insights on numerous disorders. With enhanced predictive features, transformations can be applied to each individual’s gene data, enabling more accurate decision-making compared to standard optimisations that conduct healthcare operations without the incorporation of regularisation measures. Consequently, the analytical formulation for regularisation incorporating penalty components in relation to the objective function can be articulated as follows.

Where,
Gene expression analysis
Normalisation factors are primarily employed to ensure comprehensive read counts of genes, necessitating the consideration of the overall gene length for precise measurements. Therefore, it is essential to do expression analysis for handling larger quantities of gene data with normalised components, as specified in equation (20).

Where,
Equation (20) demonstrates that substantial gene expression occurs solely when counts are normalised; thus, even with consistent representation, alterations are permitted only within specific constraints. Normalisation metrics enable the enhancement of bioinformatics data, facilitating the earlier prediction of syndromes.
Gene mapping reads
Due to the involvement of penalty variables, it is essential to establish individualised reading arrangements by mapping distinct units, hence enabling the precise alignment of gene depths and lengths. Conversely, millions of genes can be analysed if mapping points are supplied, enabling rapid predictions and classifications at each stage, as demonstrated in equation (21).

Where,
Equation (21) stipulates that for each individual unit, both the length and depth of reading configurations must be elevated, hence allowing for an increase in gene reading counts without reflecting the associated quantised indicators. Furthermore, with diminished reading capabilities, the penalty factor will be augmented, thereby allowing for the substitution of pairing indicators for each gene.
Gene batch normalisation
In response to increased biological variability, it is noted that comprehensive genetic components are altered, thus enabling the processing of batch normalisation units. Therefore, a multi-centric strategy will be employed to rectify each batch effect as specified in equation (22).

Where,
Equation (22) suggests that an increase in variations allows for a modification of the multi-centric strategy, as batch effects are diminished in this scenario. The optimisation process is illustrated as follows; block representations and flowcharts are depicted in Figures 2 and 3.

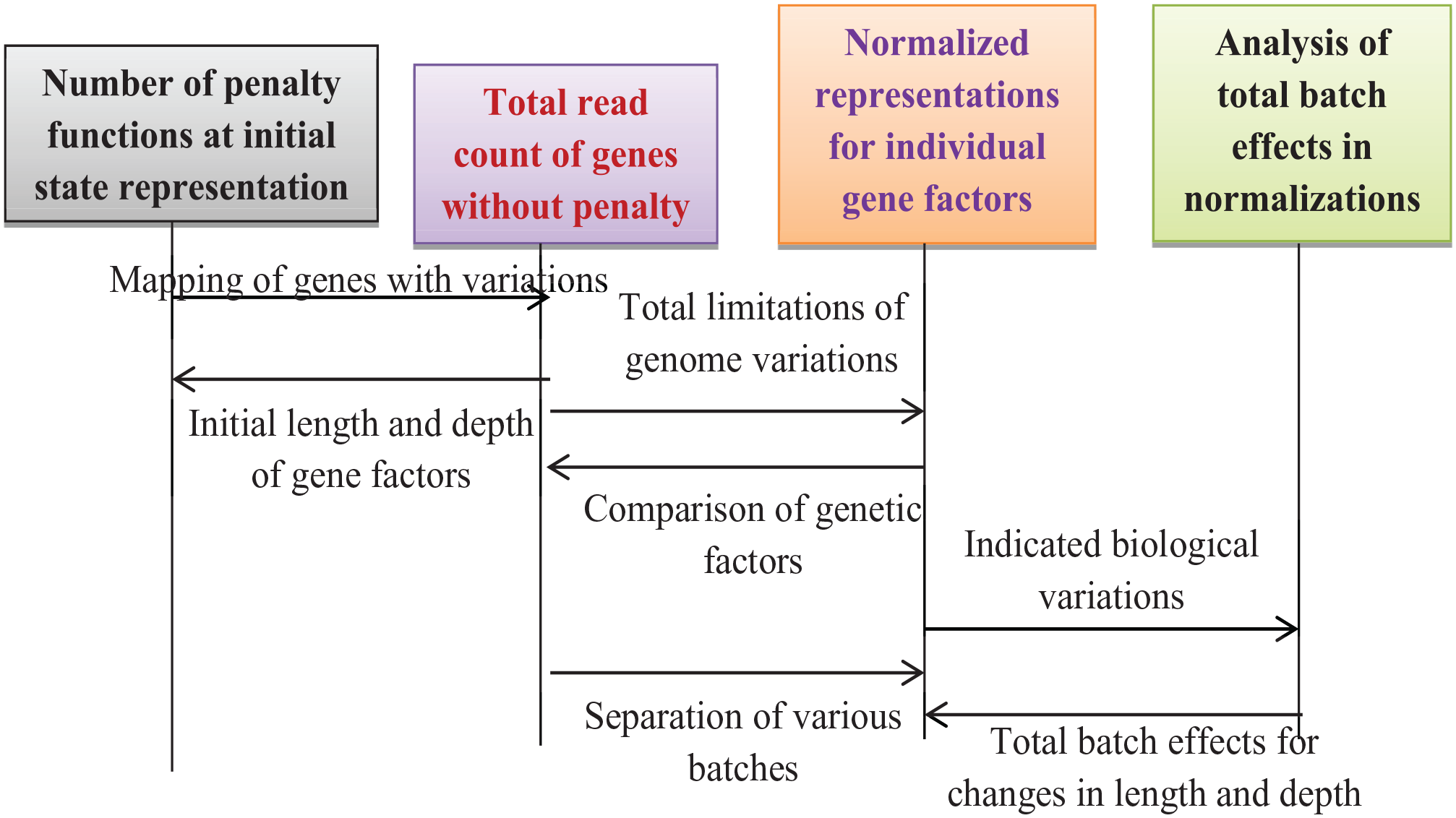
Block representations of regularisation technique in deep learning.

Flow chart of regularisation techniques in deep learning.
Hybrid deep learning optimisation
The proposed strategy, which considers various aims, will exacerbate the complexity of problems, even with the provision of normalised representations. Thus, a hybrid deep learning optimisation that amalgamates the effects of exploitation and exploration is incorporated with the meticulous fine-tuning of all requisite parameters. The primary advantage of hybrid optimisation is the establishment of a comprehensive balance among prediction, categorisation, therapy and prevention, resulting in high accuracy concerning genetic components. The flexibility of gene identification with limited data is enhanced by organising the data in sequential order. In contrast to gene matching, hybrid optimisation can find the most pertinent genes for healthcare procedures, hence enhancing the likelihood of discoveries with minimal mistake rates. Although the requisite resources exceed those of the optimisation approach that addresses singular optimised cases, scaling factors can be implemented at this stage to mitigate resource wastage. In addition to resource allocation and other relevant parameters, hybrid optimisation functions based on scheduling methods can be effectively implemented in real-time without the need for penalty functions. Consequently, in instances when individual methods exhibit limitations, hybrid optimisation can be employed to enhance speed factors, thereby reducing computing complexities. The integration of rule-based systems and data-driven models in healthcare can minimise overall computational resources, facilitating streamlined gene identification. The mathematical formulations for hybrid optimisation are as follows.
Gene matching probabilities
To achieve a comprehensive equilibrium between the original genetic component and scaled examples, it is essential to incorporate matching probabilities, with the objective of maximising the acceptance rate. Thus, the exponential case is articulated with error factors, wherein low conditional states are represented by equation (23).

Where,
Equation (23) indicates that, owing to minimal error factors, the matching probabilities can be enhanced even when additional data units are specified. Furthermore, the exploitation controls for each genetic alteration must be enhanced through the application of penalty factors, as multi-objective representations are established.
Exploration-exploitation trade-off
To provide suitable trade-off circumstances, a greater number of actions for gene indications must be selected; consequently, distinct incentives can be allocated for each action, and these indications vary according to trial factors. Thus, the trade-off between exploration and exploitation can be expressed using equation (24) as follows.

Where,
Equation (24) demonstrates that an increase in the number of activities concerning the gene factor results in an augmentation of rewards for each performance. Therefore, it is essential to preserve state-action values by minimising the similarity among different genes through the establishment of a relationship between trade-off and acquisition functions.
Genetic acquisition function
A specific gene selection point will be chosen depending on the existing solution, thereby obtaining information for efficient management. The selection of particular gene factors relies on multiple characteristics; thus, it is essential to avoid anomalies in the identification process. Consequently, only predicted values are utilised, as specified in equation (25).

Where,
Equation (25) stipulates that, while analysing exploration depth, the expected components must remain constant during all evaluated time periods. Consequently, the exploration rate escalates in scenarios where protection is unattainable, resulting in a total equilibrium with the optimisation of access probabilities. The algorithmic process of hybrid optimisation is illustrated as follows, with block representations and flow charts depicted in Figures 4 and 5.

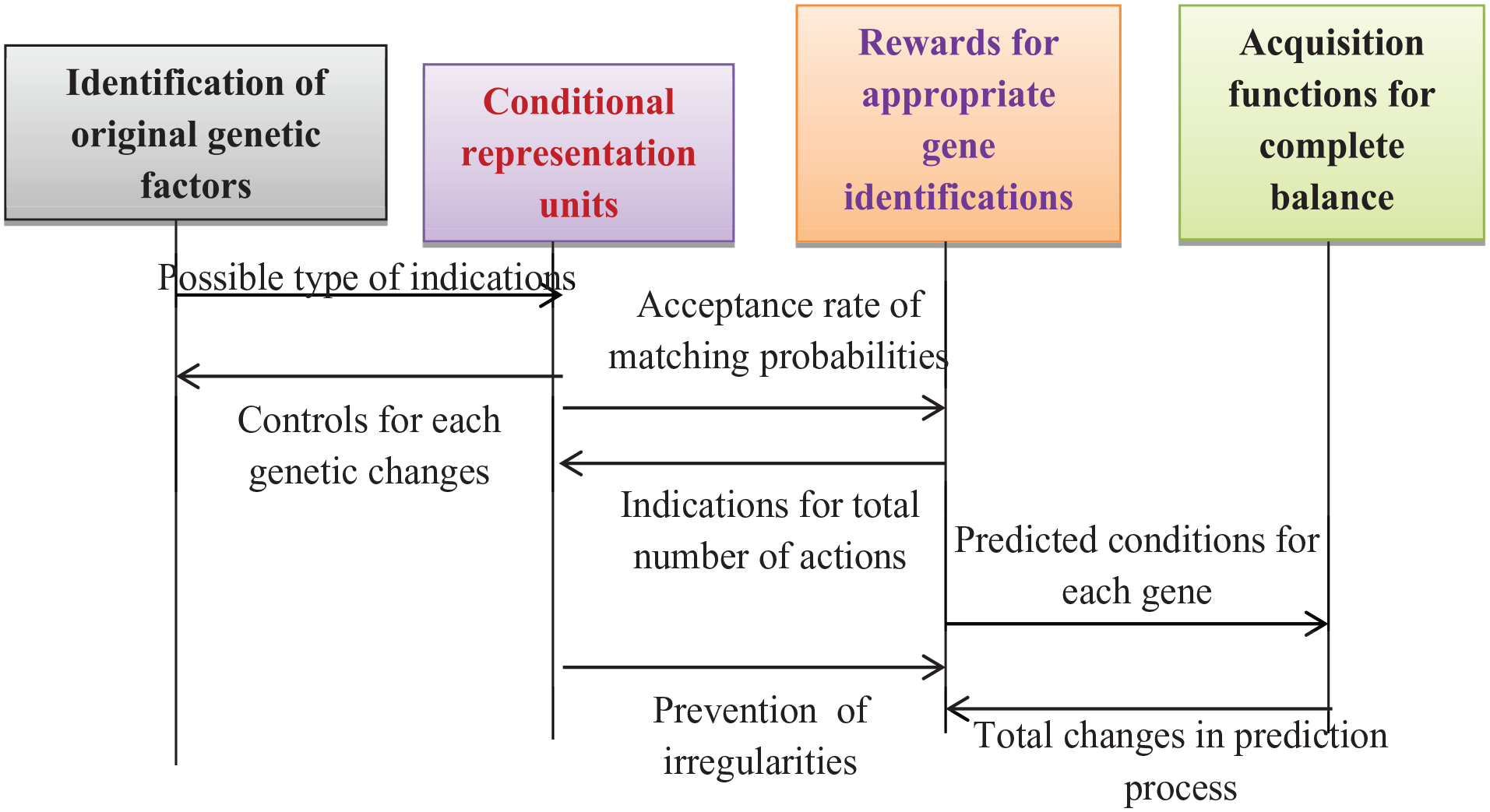
Block representations of hybrid deep learning optimisation.

Flow chart for maximisation measures with hybrid deep learning optimisation.
Results
This section examines the potential for real-time explorations by evaluating several parameters in relation to the suggested system model. Real-time outcomes must be monitored to facilitate effective healthcare operations; therefore, initial preparations are established for data collection, processing and decision-making based on dynamic interpretations. The data sources in this instance comprise genomic data, facilitating biomarker discoveries and so standardising the data effectively. In instances of irrelevant gene data, normalisation processes are implemented, altering only specified biological changes across different levels. Furthermore, comprehensive risk variables for each dataset are determined, hence enhancing the likelihood of genomic data prior to specifying numerous training criteria for each classified gene. The suggested method supplies expression factors for each genetic variant, enabling early predictions that facilitate accurate data integration and interpretation. Additionally, for enhanced biological data, cross error rates are noted, where distinct routes are identified for each gene signalling unit occupied by several users exhibiting consistent highpoints. After defining gene occupation at the corresponding rates, a cross-validation method utilising deep learning optimisation is applied exclusively to designated genetic factors, hence preventing the wastage of bioinformatics data. Conversely, to achieve real-time outcomes, a distinct bioinformatics architecture is developed, thereby enhancing the analytical robustness at each phase through hybrid optimisation, which yields balanced probability in prediction, classification, and diagnosis. To validate the results for the examined system model, 4 scenarios are analysed, and the importance of each scenario is detailed in Table 2.
Scenario 1: Representation of genomic data
Scenario 2: Probability of data integration and interpretations
Scenario 3: Minimisation of errors and data wastage
Scenario 4: Improved gain and pathway developments
Significance of scenarios.

Discussions
The results from real-time analysis are transformed into simulation-based outcomes by the accurate capture, processing and modelling of real-world data sources, so establishing a controlled hypothesis, which is detailed in this section. During the conversion phase, data from subsequent generation stages are transformed to ensure consistency among genetic data, so allowing for a consistent sequence to be maintained. In this situation, just the main features are detected by gene expression levels, so simplifying all complexity patterns with genetic dimensions in the simulation study. Furthermore, the proposed system model is constructed with an analytical representation, resulting in the creation of a regulatory gene network to facilitate the connected demonstration. Subsequently, following the representation of the simulation framework, the environmental model must be delineated, as presented in Table 3.
Simulation environment.

The conversion process serves as a secure platform for conducting genetic trials, hence facilitating improved data-driven decisions in healthcare applications. The identified gap in current methodologies can be addressed using appropriate computational techniques, such as deep learning, implemented in the suggested method. In the simulation environment, data sources are integrated with both the client and server; hence, the data collection toolbox is favoured for efficient data management. Moreover, each molecular level is delineated, allowing biological pathways to be handled within the simulation platform without external network intervention. Therefore, in the event of interventions from unspecified genetic networks, it is feasible to modify all requisite parameters, thereby enhancing gain functions. The concise overview of the contemplated scenarios is as follows.
Scenario 1: Representation of genomic data
This scenario examines the possibility of genomic data essential for accurate identification, with the objective of mitigating associated risk factors. In all healthcare procedures including prediction and classification, it is essential to normalise each genetic dataset to prevent conflicts arising from identical data sets. When identical data is present, the same genetic components are recognised, resulting in inaccurate diagnoses; hence, the suggested method evaluates the equivalent probability condition of each data set. During the testing process, any genetic data containing risk factors will be eliminated together with associated penalty factors. The reading condition in this instance is entirely enhanced as each genomic dataset is mapped by specifying the parameters of individual gene length and depth. The proposed strategy does not account for the potential mixing of genetic data with a higher risk due to real-time restrictions.
Figure 6 illustrates the comparative results for the representation of genomic data between the proposed and existing approaches. 7 Figure 6 demonstrates that the suggested strategy effectively reduces risk factors and maximises the likelihood of genomic data representations compared to the existing model. The primary reason for the reduction in risk factors is because deep learning optimisation offers a distinct method for the accurate identification of each gene, hence elucidating the genetic factors and the severity of issues throughout the mapping process. To validate the results of genomic data representations, the genetic factors analysed are 188, 214, 24, 287 and 301, with the corresponding risk factors diminished to 21%, 17%, 13%, 9% and 6%. Consequently, the percentage of genetic data associated with reduced risk factors is recorded at 37%, 41%, 44%, 48% and 51% for the existing approach, while the projected strategy shows an increase to 47%, 56%, 64%, 69% and 75%, respectively.

Genomic data representations with risk factors: (a) simulation view and (b) graphical view.
Scenario 2: Probability of data integration and interpretations
Upon initialising the genomic data, the likelihood of data integrations and interpretations is assessed in this context by examining 2 distinct types of data: physical and environmental. To further mitigate risk factors, genetic data must be efficiently integrated, enabling each mapping unit to decrease the overall biological variability. In this form of data integration, it is essential to analyse individual operations, so effectively minimising duplicate cases by precise classification. To ascertain the current state of genetic representation, it is essential to evaluate all prior genetic data, and these processing units are appropriately scaled for integration at several junctures. Furthermore, during data integration, comprehensive normalisation is essential; hence, the penalty associated with the established goal function can be eliminated, and a reward function for integration can be incorporated.
Figure 7 illustrates the likelihood of data integration and interpretations for both proposed and existing methodologies. Figure 7 clearly demonstrates that data integration is executed more effectively with the proposed method compared to the existing approach. 7 The application of multivariate principles for mitigating recognised risk factors enables the integration of data with appropriate interactive units. To validate the results of data integration and interpretation, the physical data points studied are 12, 16, 19, 22 and 25, with corresponding lowered risk factors of 18%, 15%, 11%, 7% and 4%. Therefore, to further mitigate risk factors, the percentage of integrated data is optimised to 53%, 55%, 59%, 62% and 65% for the existing approach, while the suggested method achieves increases to 61%, 66%, 70%, 75% and 78%, respectively. Therefore, through effective data integration, it is feasible to identify several genes and their corresponding management levels.
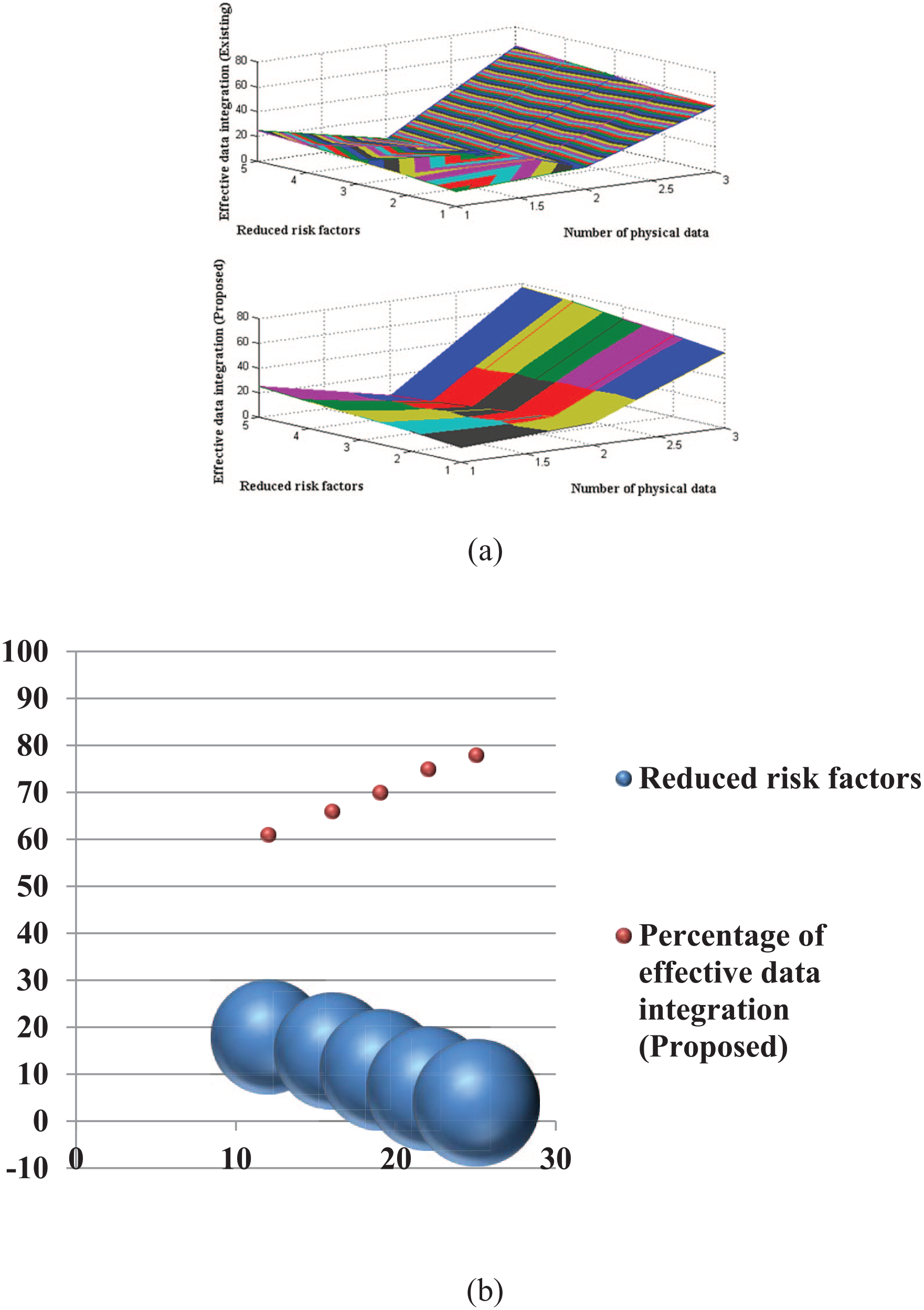
Effective data integration at reduced risk: (a) simulation study and (b) graphical study.
Scenario 3: Minimisation of errors and data wastage
This scenario involves monitoring the overall number of errors occurring during the integration of genomic data to prevent the wastage of gene data. Following data integration, the representation of numerous sets has heightened the potential for mistakes and waste, necessitating reduction through suitable regularisation techniques. Consequently, both the quantity of mandated data and the requisite amount of data are analysed, and decisions are made with adjustment factors, ensuring minimal adjustments are guaranteed. In the event of several adjustments accompanied by an increase in errors, it is essential to modify the allotted genetic data set, resulting in a greater loss of data. In the event of decreased error measurements, it is feasible to establish distinct categorisation units, allowing for adjustments in levels at this stage. Such modifications do not impact numerous sets of genetic data representations, resulting in useful observations.
Figure 8 illustrates the potential for mistake mitigation and data minimisation in both the proposed and existing methodologies. Figure 8 indicates that the overall number of errors and wastages is reduced in the suggested method compared to the present method. The primary component contributing to reductions is the appropriate use of adjustment variables in regularisation models, hence producing a superior trade-off between the genetic data set and the activities considered. The total instances of cross and fold errors in various genetic datasets are recorded as 7, 10, 14, 16 and 19, but the requisite number of genetic components is maintained at 3, 7, 11, 14 and 18. Consequently, the percentage of data wasted for the aforementioned needs is 32%, 28%, 24%, 22% and 20% using the current methodology. 7 In the instance of the proposed approach The percentage of data wastage is 17%, 12%, 8%, 4% and 2%, indicating that with little errors, total data wastage may be significantly reduced.

Data errors and wastage with genetic factors: (a) simulation view and (b) graphical view.
Scenario 4: Improved gain and pathway developments
To handle gene data, it is essential to design distinct pathways, allowing substantial bioinformatics data to be managed within a separate network without disruption. Therefore, in this scenario, the overall amount of enhanced gain at each created pathway is assessed alongside the improvement in the strength of individual networks. Changes in network decisions enable better management of genetic data, hence enhancing the accuracy of forecasts. Conversely, to attain enhanced gain, each genetic data point that is extracted from the original dataset will be diminished, thereby considering only the essential gene data pertinent to the relevant time periods. Furthermore, for the effective construction of pathways, it is essential to provide the total number of genes and their corresponding weighting functions, thereby obtaining the requisite information for accurate predictions that facilitate enhancements in coordinated actions.
Figure 9 presents the simulated results of gain and established paths for efficient genetic data management in both the current and suggested methodologies. Figure 9 indicates that the proposed strategy achieves superior gain maximisation compared to the existing model. 7 The primary cause of the gain rise is the segregation of essential genetic elements, which enhances bioinformatics networks where the requisite data undergoes further normalisation processing. To validate the results, the individual weights of the data are 50, 100, 150, 200 and 250, while the corresponding detached data, which signifies the required genetic information, is recorded as 21, 35, 56, 74 and 99. The existing technique shows percentage gains of 41%, 46%, 49%, 54% and 58%, while the new method demonstrates improvements of 67%, 74%, 79%, 84% and 89% respectively.

Percentage of gain for developed pathway: (a) simulation view and (b) graphical view.
Performance metrics
This section discusses the significance of performance analysis in evaluating bioinformatics solutions systematically, when the proposed system model is integrated with deep learning optimisation. The performance analysis is utilised to enhance the quality of healthcare operations, incorporating a substantial volume of biological data, hence facilitating an effective decision-making process. Following the identification of genetic components, prediction techniques will be implemented, and the accuracy of various tasks will be assessed using certain metrics, with scores required to exceed predetermined thresholds. Furthermore, the generalisation process, which entails the segregation of genetic material, remains highly robust; thus, the training process may be assessed by aligning the complete dataset with scalable criteria. Consequently, the suggested method includes a performance analysis, and the descriptions of the selected case studies are as follows.
Case study 1: Robustness characteristics
Case study 2: Space complexity
Case study 1: Robustness characteristics
The preliminary case studies utilising bioinformatics data, which encompasses diverse gene information, are evaluated to demonstrate their robustness, with variations noted over distinct time periods. In many instances, it is essential to analyse the growth rate, allowing for the assessment of the likelihood of increases in various data aspects, which might result in excessive robustness that should be circumvented or necessitates prior information for individuals. Furthermore, the suggested method evaluates the resilience of deep learning by correlating each biological dataset with its associated genetic information. Moreover, each genetic feature is obtained by the prediction of genetic information, hence enhancing the robustness of the stored biological data. Despite the inclusion of regularisation components, the variable level of the data must be managed; hence, the results are analysed concerning robustness features.
Figure 10 illustrates the comparative analysis of robustness between the proposed and existing approaches. 7 Figure 10 indicates that the robustness of the suggested methodology utilising deep learning optimisation is diminished in comparison to the previous method. The primary cause of diminished robustness in bioinformatics data is the aggregation of genetic data, which amplifies the trade-off at critical junctures. To validate the results of robustness characteristics, the number of iterations is incrementally increased from 10 to 100 in increments of 10, with robustness assessed at each interval of change. Consequently, the robustness for the aforementioned variations is recorded as 12, 17, 11, 15, 18, 10, 13, 16, 12 and 9 for the existing methodology, whereas for the suggested method, the robustness values are altered to 7, 10, 4, 8, 6, 7, after which only constant variations are noted.

Level of robustness for changing iteration periods.
Case study 2: Space complexity
Given the increasing issues associated with managing larger volumes of data, it is imperative to assess space complexity, which is seen as a significant computational issue, as each data element varies in relation to input size. Furthermore, the spatial complexity is assessed to determine the scalability of the data, as credible measurements must be conducted for various genetic profiles. Furthermore, for next-generation genomic sequencing, the data volume will increase; therefore, the accuracy of predictions must remain unaffected by low space complexity, necessitating the integration of multi-omics data in the suggested method. In contrast, space complexity is relevant solely for bioinformatics data that has constrained resources throughout each processing interval. The processing of individual imaging features through the integration of genetic attributes must not compromise the dataset’s structure, hence ensuring dependable breakthrough points for effective evaluations.
Figure 11 illustrates the comparative analysis of space complexity between the suggested method and the existing methodology. 7 Figure 11 illustrates that the spatial complexity of the proposed approach is diminished in comparison to the present model. The effective framing of data structures through deep learning optimisation, by organising each biological data in an appropriate sequence, enables a reduction in space complexity within the suggested model. To validate the results of space complexity, the optimal epochs are designated as 20, 40, 60, 80 and 100, during which the complexities in the existing approach are diminished to 19%, 14%, 11% and 8%. In the suggested method, space difficulties are decreased to 5.1%, and after the 40th iteration, only constant space complexities are detected, thereby preserving critical properties. Consequently, simplified processes in bioinformatics are conducted by retaining just essential information in memory storage, thereby minimising complications.

Comparison of space complexities for best epoch.
Conclusions
The suggested method primarily emphasises an optimisation framework that effectively addresses significant issues in healthcare, particularly the inefficiencies associated with big biological data. The proposed system model is created to handle all key issues by considering several parameters that encompass genetic information for processing. A primary requirement for such individual formulations is the reduction of comprehensive risk variables under equivalent probability situations, thereby addressing multiple hazards associated with both integration and interpretation. Additionally, as numerous data sets are presented, the potential for 2 distinct types of mistakes, namely cross and fold faults, is noted alongside preventive strategies. Subsequently, separate networks are established for processing each data set, using pathways that exclusively accommodate alterations in genetic information, hence enhancing the gain that signifies accuracy and classification attributes. The proposed methodology employs a hybrid technique that integrates the system model with diverse factors in alignment with deep learning optimisation, hence improving prediction outcomes.
The results of the hybrid technique are examined across 4 scenarios, and performance indicators are evaluated through 2 case studies. In the initial scenario, the risk variables are detected, resulting in 75% of genomic data, in contrast to the previous method, which yields just 51%. The proposed strategy yields a 2% reduction in data wastage while achieving around 78% data integration across various scenarios. Consequently, the gain is optimised to 89% in the projected strategy, whereas the existing model enhances the gain only to 58%. In the future, real-time explorations may be conducted via federated learning, wherein complete strategies will be assessed to transform healthcare operations.
Footnotes
Funding:
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Declaration Of Conflicting Interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
SS and HM: Conceptualization, Methodology, Software, Validation, Formal analysis, Investigation, Resources, Data Curation, Writing—original draft preparation, Writing—review and Editing, Visualization, Supervision.