
Abstract
Introduction:
The rate of acute hepatitis C increased by 7% between 2020 and 2021, after the number of cases doubled between 2014 and 2020. With the current adoption of pan-genotypic HCV therapy, there is a need for improved availability and accessibility of this therapy. However, double and triple DAA-resistant variants have been identified in genotypes 1 and 5 with resistance-associated amino acid substitutions (RAASs) in NS3/4A, NS5A, and NS5B. The role of this research was to screen for novel potential NS5B inhibitors from the cannabis compound database (CBD) using Deep Learning.
Methods:
Virtual screening of the CBD compounds was performed using a trained Graph Neural Network (GNN) deep learning model. Re-docking and conventional docking were used to validate the results for these ligands since some had rotatable bonds >10. About 31 of the top 67 hits from virtual screening and docking were selected after ADMET screening. To verify their candidacy, 6 random hits were taken for FEP/MD and Molecular Simulation Dynamics to confirm their candidacy.
Results:
The top 200 compounds from the deep learning virtual screening were selected, and the virtual screening results were validated by re-docking and conventional docking. The ADMET profiles were optimal for 31 hits. Simulated complexes indicate that these hits are likely inhibitors with suitable binding affinities and FEP energies. Phytil Diphosphate and glucaric acid were suggested as possible ligands against NS5B.
Keywords
Introduction
The primary cause of chronic hepatitis, cirrhosis, and hepatocellular carcinoma (HCC) is the hepatitis C virus (HCV). Approximately 3% of the global population have a chronic HCV infection, and for 10 to 30 years, 30% of carriers are predicted to experience significant liver-related illnesses, such as HCC. 1 The number of cases of acute hepatitis C that have been reported has increased by 129% since 2014. 2
HCV is an enveloped +ssRNA hepacivirus of the Flaviviridae family with 7 known genotypes and unknown genotypes with several subtypes that mostly affect the liver. Its 9.6 kb genome is flanked by 5′ and 3′ UTRs with a single ORF that encodes for a polyprotein with 3000 amino acids 3 and is post-translationally processed by cellular and viral proteases to yield 11 viral proteins composed of 7 nonstructural proteins (NS) which are the 2 proteins necessary for the formation of the virion (p7 and NS2), as well as 5 proteins that make up the cytoplasmic viral replication complex (NS3, NS4A, NS4B, NS5A, and NS5B), and 3 structural proteins (1 nucleocapsid protein and 2 envelope proteins). Also encoded by the core region is an alternative open reading frame protein (ARFP) or F protein, whose function is still unknown 4 even though some roles have been proposed including; regulation of Viral Translation, induction of apoptosis, interaction with host factors, and in viral morphogenesis.
NS5B is a 66 kDa heart-shaped catalytic component of the HCV replication complex, and owing to selectivity and non-toxicity, it makes a good therapeutic target due to the lack of mammalian counterparts. Like other polymerases, it has the palm, thumb, and finger domains surrounding the enzyme active site in the palm domain. 5 Many anti-HCV Direct Acting Antivirals (DAAs) that are either nucleotide inhibitors (NIs) or non-nucleotide inhibitors (NNIs) targeting NS5B are in development.6,7 At least 4 allosteric inhibitory sites have been reported (thumb site I or II, palm site I or II) 8 with their mechanisms of action detailed elsewhere.5,9 -13 However, these allosteric inhibitors have several drawbacks including low potency in enzymatic assays, 14 lack of cellular potency during an HCV sub-genomic replicon assay, 15 high lipophilic character, and low genetic barrier to resistance. 15 It is possible for distinct forms of HCV with various amino acid changes that confer treatment resistance to coexist inside the same host in the context of quasispecies. While some alterations are not linked to drug resistance, others can result in a phenotypic decrease in susceptibility to 1 or more antiviral drugs. 16 documented several substitutions with no discernible antiviral resistance to Dasabuvir and Sofosbuvir. Multi-drug-resistant RAASs variants of NS3/4A in GP1 and GP5 along with DAA-specific NS3/4A, NS5A, and NS5B were identified pan-genotypically. 17 Resistance to daclatasvir and sofosbuvir was conferred by L2003M and S2702T of NS5B, respectively. D1194A NS3/4A was triple DAA (simeprevir, faldaprevir, and asunaprevir) resistant. Double-drug resistant variants included R1181K (faldaprevir and asunaprevir), A1182V and Q1106K/R (faldaprevir and simeprevir), T1080S (faldaprevir and telaprevir), while single drug-resistant variants were V1062L (telaprevir), D1194E/T (simeprevir), D1194G (asunaprevir), S1148A/G (simeprevir), and Q1106L (Boceprevir) of NS3/4A were determined. 17 Many NS5B RAASs in genotypes 1, 3, 4, and 5 have been reported. 17 Other mutations in NS5B that were related to DAAs resistance include E237G, S282R, L320F, V321A, and V321I. 18 S282T induces high sofosbuvir-resistance, Q309R is a ribavirin-associated resistance, E237G was identified in the successfully amplified non-responder sample. 19
With a multiverse of biochemical compounds (cannabinoids and non-cannabinoids (Phenolics, Terpenes, and Alkaloids)), cannabis spp. has been reported by several communities to have medicinal activities against several disease-causing pathogens 20 including HCV. HCV Population had lower rates of diabetes and obesity when they consumed cannabis, however, whether this translates into lower mortality should be investigated. 21 It has been observed to address key challenges like nausea, and depression in HCV Patients. Some individuals receiving therapy for HCV may benefit virologically and symptomatically from modest cannabis use by helping them stick to the difficult medication regimen.22,23 Faster decay of HIV RNA among cannabis users who were HCV co-infected and reduced risk of steatosis was observed.24,25 The advantages of using cannabis for treating HCV from a biological and clinical standpoint, as well as the efficacy of this treatment, should be investigated in bigger study populations. Since there is no known vaccine or treatment for this virus, there is an urgent need for effective ways to manage and eventually eradicate this illness. This work aimed to investigate, using in-silico methodologies, if cannabis chemicals could block NS5B from HCV. Artificial intelligence (AI) was combined with traditional docking, binding energy, and simulation studies to validate AI results, quantify protein-ligand binding, and forecast the stability of the complexes. This combination significantly decreases the time and increases the accuracy needed for drug development.
Molecular dynamics (MD) simulations have become an invaluable tool in the fields of biology and drug development. These computational techniques allow researchers to study molecular systems at an atomic level over time, providing insights that are otherwise difficult to obtain through experimental approaches alone. By simulating the behavior of biomolecules, MD offers several key advantages in understanding biological processes and accelerating the drug discovery pipeline.26 -28 Molecular dynamics (MD) simulations offer critical advantages in biology and drug development. They provide atomistic insights into the behavior of biomolecules, allowing researchers to observe dynamic processes like protein folding and conformational changes, which are difficult to capture experimentally. 29 In drug discovery, it helps predict ligand binding affinities, guiding the identification of potential drug candidates by simulating interactions between drugs and target proteins. 30 Additionally, MD is valuable for studying protein stability and misfolding, shedding light on diseases like Alzheimer’s.
MD simulations also allow for the investigation of complex biological processes, such as enzyme catalysis, and help researchers explore how proteins function under different conditions, like varying solvent environments. This flexibility enhances understanding of biological mechanisms and supports the design of drugs that better target disease processes. Furthermore, MD is cost-effective and accelerates research by reducing the need for exhaustive lab experiments. These benefits make MD simulations indispensable for advancing biological research and drug development. 27
Materials and Methods
Structures
The protein structure was downloaded from pdb (PDB ID: 3FQL) while cannabis small molecules were downloaded from the Cannabis Compound Database (CBD).
Geometric deep learning virtual screening
PDBbind database 31 Protein-ligand complexes were used as inputs to train the model. Complexes that were also part of CASF-2016 32 and those that failed pre-processing likely due to errors such as incomplete structure data, missing ligand coordinates, or errors in molecular surface generation, were excluded. The remaining complexes were randomly divided into training (14 000) and test (2367) sets. The detailed representation of ligand molecules is as in Méndez-Lucio et al. 33 and the protein targets were processed using a pipeline described by Gainza et al. 34 Two separate residual graph convolutional neural networks with the same architecture, 1 for the ligand and the other for the target, were used to extract features to build the model, the extracted features were concatenated and used to build a mixed-density network (MND).
Initially, a linear layer is used to project the node and edge features to a 128-dimensional embedding. Each node and edge was updated using a series of 3 GNNs depending on the nodes that were next to them and the kinds of edges that connected them. The GNN initially updates each edge in the graph by using a multi-layer perceptron (MLP) on the concatenation of the edge features and the features of the 2 connecting nodes and the updated edge features are used to update the node feature. 33 The modified edge and node features can be utilized as input for a subsequent convolution round because they contain information about not only the core atom but also its surrounding neighbors. Three convolutions were used. The node and edge features were then processed by the remaining GNN blocks. 33
After being pairwise concatenated, the node features recovered by the GNNs and residual GNNs for the ligand and target are fed into an MND. 35 Concatenated target and ligand node information are combined to construct a hidden representation by the MND using an MLP. The MND’s outputs are computed using the hidden representation. Furthermore, by connecting neighboring nodes, the retrieved ligand node properties were utilized to forecast the type of bond and atom, aiding in the learning of molecular structures and speeding up the training. Every MLP that is utilized consists of a linear layer, and an ELU activation function. The Exponential Linear Unit (ELU) was chosen over ReLU due to its benefits in stabilizing training by allowing for smoother gradients, especially in complex networks like GNNs. ELU’s ability to output negative values helps combat the vanishing gradient problem, which can be crucial in deep architectures. Experimenting with the RELU Section has been added to future work. A dropout rate of 0.1 was employed 33 appropriate to prevent over-regularization while maintaining model generalization. The dropout rate of 0.1 was determined based on previous studies and experimentation with similar deep-learning models. Given the complexity of GNNs and the molecular nature of the data, a smaller dropout rate was appropriate to prevent over-regularization while maintaining model generalization. Experimentation confirmed that 0.1 provided the right balance between reducing overfitting and maintaining predictive accuracy.
Training
The Adam optimizer was utilized to update the model weights at a learning rate of 0.002. The loss function was minimized during model training. 16 protein-ligand complexes were used as the batch size for 150 epochs of training the model. The learning rate (0.002), batch size (16), and number of epochs (150) were primarily based on best practices from the literature on geometric deep learning models, including those for graph neural networks (GNNs). These values have demonstrated stable convergence with similar contexts, and they balance training time and performance and were refined based on preliminary runs and validation accuracy. A potential specific to a given target-ligand complex was defined using the loss function. This potential was then used to score the target-ligand complex’s 3-dimensional structure by summing over all possible pairs, calculating the negative log-likelihood for each target-ligand node pair, and calculating the distances separating each target node from each ligand node in that particular conformation. This negative log-likelihood minimizes deviations between predicted and true binding conformations, making it ideal for the drug design domain where precision in ligand positioning is crucial. The likelihood of finding the target-ligand combination in that particular conformation increases with a decreasing value.32,33
Benchmarking
The CASF-2016 benchmark, 32 which includes 285 carefully chosen protein-ligand complexes, was used to evaluate this. The preprocessing of the structures from this benchmark was identical to that of the training set. The power of screening and scoring was assessed. 33
Prediction of binding conformations
The relative location of the ligand in Euclidean space, the dihedral angles of all rotatable bonds in the molecule, and the Euler angles were used to represent the ligand conformation as a vector. Differential evolution 36 was used to determine which ligand conformation would interact with the target binding site most likely following the model, that is, minimize the potential learned by the model for that particular complex. Using a population size of 150, the global optimization was performed up to 500 iterations with a recombination constant of 0.8 and a mutation constant randomly modified from 0.5 to 1.0 in every generation. Euler angles and the dihedrals of rotatable bonds were limited to values between −π and π without seeding. 33 The detailed geometric learning protocol can be accessed in the original publication. 33 The deep neural network learns the parameters of a mixture model that is employed as a probability density function. This probability density function is used to determine the most likely distance separating a ligand atom from a specific point in the molecular surface of the binding site. The potential is determined as the combination of the negative log-likelihood of all pairwise combinations of ligand atoms and points in the molecular surface. The optimal conformation is the one that minimizes the potential, that is, the ligand conformation in which every atom is separated from the target surface by the most likely distance.
Conventional Docking
Structure preparation
The protein structure was cleaned and preprocessed by assigning bond orders using the CCd database, adding Hydrogens, creating zero bond orders, and creating disulfide bonds as well as generation of het states using Epik 37 (PH 7 ± 2 units), water, and ligands were removed and H-bond assignment was done using PROPKA in Maestro 12.8 in the protein preparation wizard. The ligands were prepared by Ligprep. 38 Briefly, the ligands were imported into the maestro workspace, and only those with a maximum of 300 atoms were subjected to OPLS4 Force force field, 39 and ionization was done using Epik 37 to generate possible states at PH = 7±2 units.
Glide docking
The prepared structures were subjected to XP Glide 40 with only ligands of atoms and rotatable bonds equal to or below 300 and 100 respectively were selected with a Vander Waals scaling factor of 0.8 and a partial charge cutoff of 0.15. The receptor was rigid with flexible ligand sampling and to sample nitrogen inversions and ring conformations. Bias sampling torsion was set for all predefined functional groups and Epik state penalties were added to docking scores. Post-docking minimization was performed with 10 poses per ligand with a threshold of rejecting minimized pose set to 0.5 kcal/mol.
Binding affinity determination
SeeSAR 12.1.0 41 was used to perform binding affinity calculations. A binding site was defined by the co-crystallized ligand in the receptor PDB file and copied to the docking mode after the generated protein and docking library were loaded into the Biosolveit workspace. 41 Docking calculations were done for each compound in a standard docking mode with defaulted settings and parameters. The affinity of the generated poses was then assessed and the best poses were selected based on these affinities.
Binding energy is calculated from the HYDE score function (equation (1)), which relies on intrinsically balanced terms of atom-specific desolvation, hydration, and hydrogen bonds based on the logP atomic increment system, 42 while also taking into account the Torsion angle values to the binding conformation of the protein-ligand. The quotient of G and the number of non-hydrogen atoms in the molecule is used to define a ligand’s binding affinity (equation (2)) 43 and expressed as.
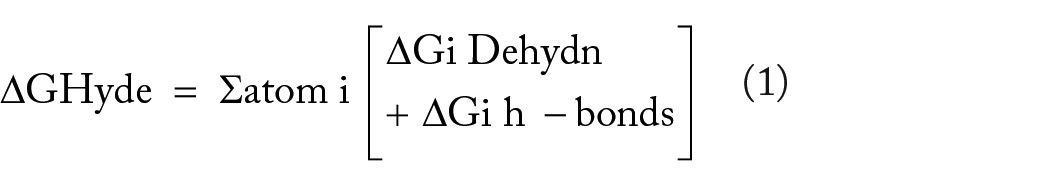

where ΔG = −RT ln Ki and N = number of non-hydrogen atoms.
The selection of the best poses was based on their visual HYDE scores while also considering a statistics-based torsional analysis.
ADMET screening
All compounds were subjected to SeeSAR, 41 AdmetLab2.0, 44 and QikProp.29,45 The protocol used for SeeSar is as in the Binding Energy determination above. The concatenated sdf file was uploaded to the AdmetLab2.0 website (https://admetmesh.scbdd.com/) in the ADMET screening mode. It employs a Multi-task Graph Attention (MGA) framework made up of input, Relation graph convolution network (RGCN) layers, attention layer, and fully connected (FC) layers.
Molecular simulation dynamics
All simulations were done in GROMACS 2021.4. 46 The protein topology was generated using AMBER99SB 47 force field from pdb2gmx module while ligands were parametrized by using Generalized Amber Force Field (GAFF2) 48 at antechamber website. The complexes were placed in the hexahedral box using gmx editconf, solvation was done using gmx solvate, TIP3P water molecules were added to the systems, and Na+ and Cl− ions were added using genion to a concentration of 0.15 m. Equilibration was done with steps set to 20 000 in 10 ns at 298 K and 1 atm with position restraints Force Constant set to 700 kJ/mol. This allowed water molecules and ions to move freely. Production MD simulations lasted for 100 ns and they were monitored by checking system energies during the simulations. Pymol, vmd, and Gromacs binaries 46 were used for the analysis of the results.
MM/G(P)BSA calculations
Three energetic terms are taken into account in the computation of binding energy when determining the free energy of complex formation in conjunction with MD simulations: (1) variations in the system’s potential energy in a vacuum; (2) polar and apolar solvation of the various species; and (3) the entropy related to complex development during the gaseous phase.
The Uni-GBSA-based tool unigbsa-traj was utilized to do MM/PBSA (Molecular Mechanics/Poisson-Boltzmann Surface Area) calculations automatically for every simulated system.45,49 The formula for estimation of free energy is explained in detail in Charles et al, 45 Wang et al, 50 and Gilson and Honig. 51
Free energy perturbation (FEP) calculations
The CHARMM-GUI Free Energy Calculator and NAnoscale Molecular Dynamics (NAMD) were used to calculate absolute free energy.29,52,53 The simulated ligands (docked postures) were uploaded as a single concatenated SDF file, and CHARMM General Force Field (CGenFF v1.x) 54 was used for ligand parametrization and topology construction to build the NAMD inputs and post-processing scripts. Counter ions (KCl) were used to neutralize all of the systems that were collected to produce input files and post-process scripts. Applying restraining potentials to limit the ligand’s location in a receptor during FEP/MD allowed for the calculation of binding free energy using the double decoupling approach. With the resulting inputs, the TIP3P water model and Langevin piston pressure were applied to the system’s NPT ensemble at 300 K and 1 atm. The FE values were captured in history files obtained via FEP lambda replica exchange MD (λ-REMD) using simple overlap sampling (SOS). By measuring the FE values throughout the last 6 ns, the final FE values from 10 ns FEP/λ-REMD simulations were computed. 29 The sequence of the used methods is shown in Figure 1.

The sequence of the steps followed in this study.
Results and Discussion
Geometric deep learning virtual screening
The results show that all optimal binding conformations had negative values (Figure 2 and Table 1) and only the best 100 molecules were considered for further studies based on their potential scores, with malic acid having the best score of −286.78, however, the complexes of those ligands with many rotatable bonds (⩽10) could not achieve successful optimizations and terminated after 500 iterations (Figure 2), this is because an increase in rotatable bonds is associated with the inefficiency of the optimization algorithms when working with a large number of degrees of freedom. These ligands justified the need for conventional docking methods integration, all compounds having rotatable bonds above 10 underwent the same re-docking process 55 and average potential scores were reported in Table 1.

The results of geometric deep learning showing the distribution of some key parameters. Fun: statistical potential, nit: number of iterations, rmsd: root mean square deviation.
The summary of the results of virtual screening, molecular docking, and ADMET studies. Fun: A potential specific to a given target-ligand complex, SA Score: Synthetic Accessibility Score, LC50FM: 96-h fathead minnow LC50, FDAMDD: maximum recommended daily dose, Ames: Mutagenicity test, SR-p53: Probability of being p53 actives.

This compound has an ester and may undergo hydrolysis at high or low pH.
This compound contains an acetal/aminal-like group (X-CH(R)-Y where X, Y are N, S, or O) that may be acid/base labile, releasing an aldehyde.
Both ester and acetal/aminal-like groups.
Rotatable bonds above 10.
PAINS alert (1).
Conventional docking and binding affinity determination
The results of docking and binding affinity determination are shown in Table 1 and they agree with the results of geometric learning, validating the compounds under investigation. Phytyl diphosphate had the best score of −12.275 kcal/mol, but iso-citric acid had the best ligand efficiency of −3.100. The compounds had acceptable values of Binding affinities 41 with Isovitexin having the highest value of 619 407 to 61 541 783 nM (Table 1).
ADMET screening
The results for some important selected properties are presented in Table 1, and the summary of all properties is shown in Figure 3 and Supplemental File 2, because the presence of PAINS alert is not enough to justify the elimination of a hit candidate, 45 Compounds with pain alerts were only eliminated if at least other 4 ADMET properties were out of range (except the number of bonds, which was not considered as an ADMET property). The Synthetic Accessibility Score (SA Score) quantifies how simple it is to synthesize drug-like compounds. A score of less than 6 indicates that the compound is simple to synthesize.29,44 An estimate of the hazardous dosage threshold of substances in humans can be obtained from the maximum recommended daily dose (FDAMDD). The likelihood of being toxic is the output value, and it ranges from 0 to 0.3 for excellent (less likely to be harmful); 0.3 to 0.7 for medium; and 0.7 to 1.0 for poor (more likely to be toxic). 44 The mutagenicity test is the Ames test. The most used test for determining a compound’s mutagenicity is the mutagenic effect, which closely correlates with carcinogenicity. The numbers indicate the probability of being harmful; 0 to .3 indicates excellent (less likely to be mutagenic), .3 to .7 indicates medium, and .7 to 1.0 indicates poor (more likely mutagenic). 44 A fathead minnow LC50FM is defined as the concentration of the test chemical in water which results in 50% of the minnows dying after 96 hours. For LC50FM, the unit is −log10[(mg/L)/(1000 × MW)]. 44 One reliable sign of DNA damage and other cellular stressors is the activation of p53. The output values of SR-p53 are the probabilities of being active, with 0 to 0.3 representing excellent (likely inactive), 0.3 to 0.7 representing medium, and 0.7 to 1.0 representing poor (most likely active). 44 The rest of the computed ADMET properties are presented in Supplementary File 241,44 and their tSNE distribution is in Figure S1.
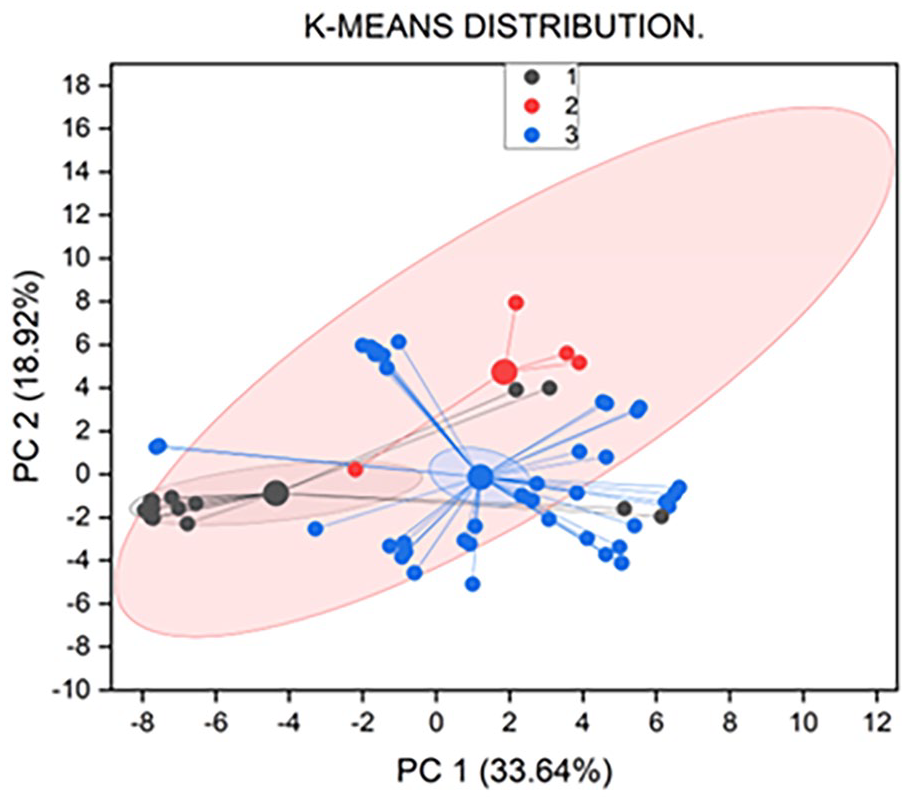
The K-means cluster analysis of ADMET properties.
Molecular dynamics simulation studies
The potential of 6 protein-ligand complexes as NS5B inhibitors was demonstrated by the formation of stable complexes by each of the simulated complexes. The analysis of Root Mean Square Deviation (RMSD) evaluates the long-term structural stability of biomolecular simulations. It estimates the typical difference between an atom’s location in a molecular structure and its reference structure.29,56 The RMSDs of each system show that during the experiment, every simulated complex reached stability (Figure 4A and S2A). Comparatively, the glucaric acid complex was more stable than the other compounds. The observed RMSD differences between the complexes and their corresponding proteins exhibit a variation that is driven by the ligand. When utilizing 1D RMSD, it is easy to believe that 2 structures that have the same RMSD from a reference frame are similar, but in practice, they can differ significantly. Alternatively, significantly more information can be obtained by computing the RMSD of each frame in the trajectory to all other frames in the other trajectory to give 2D RMSD. 45 Pairwise RMSDs of each trajectory were calculated to itself and the results are shown in Figure S5, the diagonal represents 0 (RMSD of a structure to its self), and all the structures had RMSDs ⩽ 1.8 Å, over simulation showing that the complexes were stable over simulation and had nearly same states but not identical except Phytyl diphosphate complex had more re-visited states than other complexes (Figure S5). These results agree with 1D RMSD results and show that the glucaric acid complex was more stable evidenced by lower RMSD values.

The molecular dynamics simulation results: (A) illustrates the RMSD variation for proteins across different complexes, (B) displays the total interaction energy for each system throughout the simulation, (C) shows the RMSF variation over time, and (D) presents the changes in the radius of gyration (Rg) during the simulation.
Root Mean Square Fluctuation (RMSF) analysis is another way to understand the flexibility and dynamic behavior of individual atoms or residues within a biomolecular system as well as their contribution to the flexibility of the whole molecule.26,30,57 The main interacting residues have minimum RMSF values during simulation time compared to the ligand-free protein, supporting their stability and interactions with the simulated compounds, while the RMSF of non-interacting residues shows somewhat greater oscillations (Figure 4C). The distribution of the rmsf further supports the stability of all complexes (Figure S4D).
The Radius of Gyration (Rg) is a measure of the compactness or spread of a biomolecular structure in 3-dimensional space is valuable for analyzing the overall shape and structural fluctuations of biomolecules.30,45 A relatively constant Rg value fundamentally signifies a stably folded structure and a reduction in Rg signifies an increase in stability.29,30,45 All of the simulated systems had Rg values between 23.7 and 24.2 Å, indicating their stability. The simulation revealed a fairly progressive reduction in the gyration radius over time, indicating a gradual increase in the compactness and stability of all systems (Figure 4D). The distribution of Rg also shows that the Glucaric acid complex is more stable relative to other complexes (Figure S3C).
Solvent Accessible Surface Area (SASA) is a measure of the surface area of a biomolecule that is accessible to solvent molecules. 58 It plays a crucial role in analyzing the interactions between biomolecules and their surrounding solvent environment. 45 A higher value suggests an increase in the protein’s volume, indicating lower stability, whereas stable proteins typically exhibit minimal fluctuation throughout the simulation. The binding of a small molecule can alter the solvent-accessible surface area (SASA) and significantly impact the protein’s structure.28,45,59 The SASA consistently decreased across all systems during the 100 ns simulation (Figure S2B). This decrease in SASA indicates a rise in compactness, consequently indicating enhanced stability across all systems. The parallel patterns observed in both SASA and Rg affirm the validity of the molecular dynamics simulation outcomes. 45 Their distribution also shows that all systems were stable over simulation with the glucaric acid complex being more stable than others (Figure S3C and S3D).
Non-bonded Molecular Mechanics (MM) interaction energy between the Ligands and their receptor was calculated to evaluate the magnitude of the interaction between the ligand and the protein.60,61 The total interaction energy for all the systems was negative over 100 ns simulation with the complex of phytyl-diphosphate having the lowest energy values (Figure 4B and S3B). Vander Waals forces are short-range interactions that include London dispersion forces and dipole-dipole interactions, they enable the close interaction between the nonpolar regions of the ligand and the protein, ensure complex shape complementarity, predict the strength of the protein-ligand interaction with higher energy indicating stronger binding. 62 Apigenin coumaryl glucoside had the best values while that of Isocitric acid was the worst, a fact justified by their size and proximity to the protein groups (Figure S9). The stability of the complex and total binding energy can be enhanced by the long-range electrostatic interactions, which can direct protein molecules toward their pre-binding orientations. 63 Phytyl Diphosphate had the best values of electrostatic energy while apigenin coumaryl glucoside had the worst values since the former has phosphate groups which results in strong electrostatic interaction energy. These energies are good predictions of binding affinity since they are considered while computing those affinities. The simulated complexes remained stable throughout the simulation according to this data (Figure S2C and S2D). But it’s crucial to remember that this quantity is neither a binding energy nor a free energy. 60 Figure 5 displays the specifics of the molecular interactions that occur in the middle of the simulation. The summary and distribution of all these energy terms are shown in Figures S3B, S4A, S4B, and S4C.

An overview of the interactions between the selected compounds and the target protein. The interacting residues and compounds are shown in licorice representation, with the residues highlighted in green and the compounds in pink, while the rest of the protein is displayed in a cartoon format.
During the simulation, the minimum distances between active site residues and ligands were calculated. Since most of the time, these minimum distances were less than 3 Å, a conventional hydrogen bond can still form as long as the acceptor and donor are orientated correctly (Figure S3A).
PCA (Principal Component Analysis)27,64,65 was performed particularly on alpha carbon data from the last 25 ns to acquire insights into the dynamic behavior of both the complexes, incorporating structural and energy data. This analysis aimed to explore the conformational range of the complexes, distinguishing various regions within the energy landscape explored during the MD simulation. The complexes defined discrete conformational clusters and suggested stability by occupying compact subspaces. A graph illustrating the motion and displacement of atomic fluctuations within the complexes was created by utilizing eigenvectors 1 and 2. The first few eigenvalues had greater values, and the remaining eigenvalues were in a declining order. 45 All complexes showed PC ranging from ~15 to ~−15 (Figure S6). The simulation results of the apo-protein are shown in Figure S10. Interacting residues have increased positive correlation, as indicated by the Dynamic Cross-Correlation (DCC) data, indicating that they are interacting with the target. Residues located in the active site showed slightly elevated positive cross-correlation peaks upon ligand binding, suggesting a high occupant binding affinity (Figure S8). 45
The average binding free energy of simulated hits was estimated using MM/P/GBSA calculations, and the outcomes are in good agreement with other studies. Since all energies were negative, proteins and ligands were strongly bound together (Table 2). Additionally, FEP/MD simulations were performed; Table 2’s results show that these ligands have sufficient binding affinities, making them eligible for in-vitro research. To evaluate the convergence and dependability of the findings, the FE values were averaged using the standard error of the mean (SOS). The SEM of all data is less than 0.5 kcal/mol, indicating that all systems have converged in less than 10 ns.29,52,66
The table shows the results of MM_G/PBSA and FEP/MD energy calculations.

Because of many RAASs in HCV proteins, reported DAAs resistance-enhancing RAASs were introduced in this protein with several combinations as shown in Table S1 and these mutants were re-docked with the identified compounds. 7 ligands still had significant docking scores with Glucaric acid having the highest values (Figure 6 and Table S2). Paired two-tailed t-tests to compare the wild type and mutants are also shown in Tables S4, S5, S6, and S7. These results show that the docking scores of many ligands are significantly reduced except for the 7 ligands. This may suggest that some of these compounds, with glucaric acid leading, may still bind the resistant phenotypes. While these docking results provide valuable insights, they alone are insufficient to fully demonstrate the efficacy of the identified ligands against the mutant proteins. Additional investigations, such as molecular dynamics simulations of the mutant complexes, will be necessary to further validate and understand their interactions and potential effectiveness.26,57,64,67

The results of docking the ligands into the active sites of mutants.
Studies have demonstrated the hepatoprotective, anti-inflammatory, cholesterol-lowering, anti-oxidant, and anti-carcinogenic properties of glucaric acid and its derivative D-saccharic acid-1,4-lactone. 68
With the pan genomic approach to HCV therapy, the candidates were also docked with NS3/4 protease (PDB ID: 3P8N), and the results are presented in Table S3, 4 ligands including Glucaric acid had docking scores better than −7.00 kcal/mol. Having inhibitors inhibiting both proteins will be a bonus for drug development. To further discover the global therapeutic world of glucaric acid, insight into the compound’s potential pharmacological targets and associated biological pathways was determined. This was done using Swiss Target Prediction to predict other possible targets of Glucaric Acid. Results (Figure 7) show that this compound may be active against several targets including the Neuronal acetylcholine receptor protein alpha-7 subunit (Probability = .765) and Squalene synthetase (Probability = .103). Inhibiting the former means that this compound can also be a competitive antagonist at a neuromuscular junction and hence can competitively compete with bungarotoxin minimizing its blockade activities at the junction 69 while inhibiting the latter is a potential therapeutic strategy for lowering cholesterol levels in individuals with hypercholesterolemia, as well as Anti-Cancer Potential Since cholesterol, is essential for the formation of lipid rafts and cell signaling pathways involved in cancer cell proliferation and survival, inhibiting squalene synthase can potentially inhibit tumor growth and metastasis.68,70 The identified hits are shown in Figure S11.

The results show other possible targets for glucaric acid.
Some studies have also highlighted cannabis-delivered phytochemicals to inhibit this protein, 1 study highlights the use of terpenes, naturally occurring compounds found in plants including cannabis, as potential HCV NS5B polymerase inhibitors. The study identified several terpenes, such as mulberrofuran G, cochlearine A, and pawhuskin B, that demonstrated stable binding interactions with the NS5B enzyme. These terpenes were suggested as promising candidates for inhibiting viral replication, showing better docking results compared to traditional HCV drugs like sofosbuvir , 71 based on the distribution of molecular simulation parameters of complexes, our complexes show more stability. Additionally, another study explored various phytochemicals, including gallic acid, catechin, resveratrol, apigenin, and silibinin, which have antiviral properties. These compounds were docked against the HCV NS5B enzyme, with silibinin showing the best binding affinity, suggesting it could be a potent antiviral agent against HCV , 72 the compounds evaluated in this study exhibit superior docking scores and binding affinity ranges compared to compounds above from. 72 These results emphasize the strong potential of the studied compounds for effective target interaction, with simulations further supporting their viability, unlike the lower-performing compounds from previous studies, which lack binding affinity data and simulated validation (Table 3).
Comparison of docking scores, binding affinity ranges, and simulation data between compounds from this study and those from Shakya. 72 The compounds from this study demonstrate stronger docking scores, broader binding affinity ranges, and simulation validation, highlighting their superior potential for target interaction.

Future work and limitations
Molecular dynamics simulations will be essential to further investigate the impact of NS5B mutations at the molecular level, as well as studying these proteins under different solvent conditions, such as urea. While the docking results provide useful preliminary insights, they are insufficient to confirm the efficacy of the identified ligands against mutant proteins.65,73,74 Additional studies, including molecular dynamics simulations of the mutant complexes, will be necessary to validate these findings and better understand ligand-protein interactions for potential therapeutic applications. 26 Molecular dynamics (MD) simulations and in silico methods, including docking and AI models, offer powerful tools for studying biological systems and drug discovery, but they come with limitations. MD simulations are computationally expensive, especially for large systems or long timescales, requiring significant resources. The accuracy of these simulations depends on force fields, which may not always represent molecular interactions accurately. Additionally, simulations often oversimplify complex biological environments, leading to less realistic results. Moreover, in silico findings require experimental validation to ensure their relevance. These methods are most effective when used alongside laboratory experiments to complement and verify computational predictions.75,76 More hyperparameter tuning as well as experimenting with ReLU will be a future optimization step to see if it improves the model’s convergence.
Conclusion
We have used geometric deep learning to screen for possible HCV inhibitors of NS5B from cannabis sativa natural compounds. These compounds’ data show that they may be potential DAAs against HCV. It is however very important to keep the RAASs in check, given that they represent the biggest challenge to HCV treatments, and screening them and recommending treatment post their identification will be a step ahead. The best hits identified were Glucaric acid and phytyl diphosphate, with the former retaining the ability to interact with mutants studied as well NS3/4 hence a possibility of polypharmacology and better than the used standard of sofosbuvir.
This work broadens the scope of cannabis research by shedding light on the potential therapeutic applications of the plant beyond its well-known effects, improving our understanding of its pharmacological properties, and highlighting its potential as a source of novel antiviral compounds.
This study further guides in vitro and in vivo experimental studies for validation of the actual efficacy, safety, and inhibitory mechanisms of these compounds. Cannabis-derived compounds may offer advantages such as natural sourcing, potentially fewer side effects, and diverse chemical structures for drug optimization and could also inform the design of more potent and selective HCV inhibitors.
Supplemental Material
sj-docx-1-bec-10.1177_11795972241306881 – Supplemental material for Geometric Deep learning Prioritization and Validation of Cannabis Phytochemicals as Anti-HCV Non-nucleoside Direct-acting Inhibitors
Supplemental material, sj-docx-1-bec-10.1177_11795972241306881 for Geometric Deep learning Prioritization and Validation of Cannabis Phytochemicals as Anti-HCV Non-nucleoside Direct-acting Inhibitors by Ssemuyiga Charles and Mulumba Pius Edgar in Biomedical Engineering and Computational Biology
Supplemental Material
sj-xlsx-2-bec-10.1177_11795972241306881 – Supplemental material for Geometric Deep learning Prioritization and Validation of Cannabis Phytochemicals as Anti-HCV Non-nucleoside Direct-acting Inhibitors
Supplemental material, sj-xlsx-2-bec-10.1177_11795972241306881 for Geometric Deep learning Prioritization and Validation of Cannabis Phytochemicals as Anti-HCV Non-nucleoside Direct-acting Inhibitors by Ssemuyiga Charles and Mulumba Pius Edgar in Biomedical Engineering and Computational Biology
Footnotes
Funding:
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The authors received a grant (PBF/2023/0002) from PharmaQsar Bioinformatics Firm, Kampala, Uganda to execute this work.
Declaration of Conflicting Interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
Data Availability Statement
The corresponding author has the data supporting this work, which can be shared upon inquiry.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.