
Abstract
Schistosomiasis, which causes over 200 000 deaths annually, has since the 1970s been controlled by praziquintel. The reliance on a single drug to combat schistosomiasis, and reports of laboratory resistance to the drug, has created an urgent need in the scientific community to develop new chemotherapies to complement or supplement praziquantel. Medicinal plants are a potential reservoir of compounds with schistosomicidal activity. In the current study, we carried out computer-aided screening of Abrus precatorius compounds to discover compounds with potential to inhibit Schistosoma mansoni purine nucleoside phosphorylase (SmPNP). Thus, 99 compounds retrieved from Lotus Natural Compounds Database were docked into the active site of SmPNP. The top-ranked compounds were subjected to Lipinski’s druglikeness and toxicity risk predictions. Three lead compounds, abrusogenin, cirsimaritin and hispidulin, were identified as having high binding affinities, favourable interactions with SmPNP active site residues and good toxicity risk prediction results. Molecular dynamics (MD) simulations were used to assess the stability of the interactions of these lead compounds with SmPNP. Collectively, analyses of the MD trajectories confirms that the lead compounds bound and interacted stably with active site residues of SmPNP. We conclude that abrusogenin, cirsimaritin and hispidulin could serve as hit compounds for the development of new antischistosomal drugs, based on plant-derived natural products. However, experimental studies are required to further evaluate the potentials of these compounds as possible therapeutics against schistosomiasis.
Keywords
Introduction
Schistosomiasis is a parasitic disease caused by blood flukes (class Trematoda) of the genus Schistosoma. 1 The 6 species responsible for morbidity are Schistosoma mansoni, S. japonicum, S. haematobium, S. mekongi, S. guineensis and S. interlacum. 1 This neglected tropical disease (NTD) occurs mostly in developing countries with sub-Saharan Africa accounting for about 93% of the world reported cases.2,3 The most prevalent species in sub-Saharan Africa are S. mansoni and S. haematobium. 4 To date, Schistosomiasis treatment relies almost exclusively on praziquantel (PZQ) and oxamniquine (OXA).5-7 Other drugs such as hycanthone and lucanthone have been used, but they showed serious side effects. 7 Reports of treatment failures with PZQ and OXA due to resistance or tolerance7-10 make over-reliance on these 2 drugs to treat the disease pose a potential public health threat. There is, therefore, an urgent need for the search for potential novel antischistosomal drug candidates.
Natural product (NP)-based drug discovery research has been practiced for a long time and up to 50% of all approved drugs are considered NP, or at least were influenced by them.11,12 This is not surprising, considering the great structural diversity of NPs that make them an outstanding source of novel molecular scaffolds in drug discovery.13,14 Several plants have been investigated for their anti-schistosomal activity, both in vivo and in vitro. These plants have shown varying degrees of efficacy. Some plants that were studied include, Zizinger officinale, 15 Artemisia annua, 16 Rauwolfia vomitoria, 17 Pulsatilla chinensis, 18 Anonidium mannii, 19 and some Cucurbita cultivars. 20
In some parts of Southern Africa, Abrus precatorius extracts are used in the treatment of Bilharzia. 21 In vitro studies also suggested that extracts from the plant were lethal to adult schistosomes. 22 A. precatorius is a subtropical weedy high-climbing, twining, or trailing woody vine with leaves known to be sweet-tasting. 23 Seeds of the plant are considered poisonous and contain such toxic compounds as Abrin that has an estimated human fatal dose of 0.1 to 1 µg/kg. 24
Identification of drug candidates via computational screening approaches is a promising and cost-effective strategy that plays an important part in the drug discovery and development pipeline.25-27 In the current work, we used computer-aided approaches to screen A. precatorius compounds as potential inhibitors of the S. mansoni purine nucleoside phosphorylase (SmPNP). PNP, also known as inosine phosphorylase, is essential for the maintenance of proper cellular function and metabolism by participating both in the de novo purine synthesis and the purine salvage pathway.28,29 S. mansoni depends exclusively on the salvage pathway for its purine requirements, 30 thereby making SmPNP a promising target for the development of antischistosomal drugs.
We screened a library of 99 A. precatorius using molecular docking against SmPNP followed by in silico druglikeness and toxicity evaluations. Our results suggest that abrusogenin, hispidulin and cirsimaritin can be a potential potent drugs against schistosomiasis. The stability of the complexes of these compounds within the SmPNP active site was confirmed by Molecular Mechanics/Poisson-Boltzmann (Generalized Born) surface area (MM/PB(GB)SA) and Molecular Dynamics (MD) simulations.
Materials and Methods
The schematic representation of the methodology employed in this study is shown in Figure 1.

A summary of the experimental design employed in the current study.
Computational tools
All software were run on an HP Z240 Tower Workstation Intel(R) Core(TM) i7-6700 CPU @ 3.40 GHz, 32 GB RAM, and 915 GB hard disc space. Windows 10 Professional was the Computer Operating System.
Ligand preparation
First, the structures of the 99 A. precatorius compounds were downloaded from The Lotus Natural Compounds Database. 31 Then, the simplified molecular input line entry system (SMILES) for each compound was used to retrieve the corresponding 3-dimensional structures of the compounds from the PubChem database (https://pubchem.ncbi.nlm.nih.gov/) 32 in the simple data format (SDF). Structures in the SDF format were loaded into the Open Babel 33 plugin of Python Prescription (PyRx) v.0.8 34 for energy minimization. Energy minimization was achieved using the Universal Force Field, and the algorithm set to conjugate gradient and 200 steps. The energy-minimized ligands were then saved in the PDBQT format in preparation for molecular docking.
Receptor preparation
The 3D crystal structures of SmPNP (PDB ID: 3FAZ) was retrieved from the Protein Data Bank (PDB) (www.pdb.org/pdb) and prepared for docking using the Dock Preparation (dock prep) tool of UCSF-Chimera V 1.16. 35 Before Dock Prep, the protein was cleaned of previously bound ligands. For ease of docking calculations, only Chain A was used for this in silico study. Dock prep refinement steps included removal of water molecules, addition of missing hydrogen atoms and addition of polar charges. The prepared receptor in the PDB file format was loaded in PyRx and subsequently converted to the PDBQT file format for molecular docking.
Molecular docking
To observe the best binding poses of A. precatorius compounds within the binding pocket of SmPNP , molecular docking was performed by using AutoDock Vina software 36 within the PyRx v 0.8 platform (available at https://pyrx.sourceforge.io/). The ligand binding sites were determined using the DeepSite webserver (https://www.playmolecule.com/deepsite/) 37 and the grid box was set as X = −2.11, Y = 2.56, and Z = 28.25 with the dimensions of the grid box being (X,Y,Z) 26.82 Å × 28.38 Å × 46.43 Å. The exhaustiveness was set to the default 8. Once the molecular docking process was completed, the best pose with minimum binding affinity and zero root-mean-square deviation (RMSD) was selected for each ligand. The interactions between the docked protein and the respective ligands were visualized using LigPlot+ 38 and PyMOL (http://www.pymol.org/pymol).
Validation of molecular docking accuracy
To validate the molecular docking protocol, inosine, originally co-crystallized within the SmPNP binding pocket, was re-docked using Autodock Vina following the same procedure as already described for A. precatorius ligands. The docked pose was then superimposed to the original pose present in the co-crystallized structure using the BIOVIA Discovery Studio Visualiser v2021 software (https://discover.3ds.com/discovery-studio-visualizer-download).
Drug-likeness analysis
The druglikeness predictions of the A. precatorius compounds was done by applying Lipinski et al’s rule of five (RO5) parameters. 39 In the current study, the druglikeness properties of the compounds were calculated using Osiris DataWarrior v.4.5.1. 40 In addition to Lipinski RO5, the PAINS-Remover 41 web tool was used to filter out the pain-assay interference compounds (PAINS). 42
Toxicity risk prediction and drug score calculation
The OSIRIS Property Explorer 43 application was used to predict the compounds’ toxicity risk profiles and for calculating the drug scores of the compounds.
Re-validation of docking scores and receptor-ligand interactions
Re-validation of the binding scores and protein-ligand interactions for top ranking compounds was done by BINDSURF Achilles blind docking server (http://bio-hpc.eu/software/blind-docking-server/). 44 Briefly, BINDSURF carries out an exhaustive series of docking simulations of the ligand to the entire protein surface to identify sites with the best binding affinities. The tool then applies a pose clustering algorithm to generate clusters of the results, and the pose with the best binding affinity in each cluster is chosen as the representative. In this work, the highest scoring receptor-ligand complexes were visualized and analysed using LigPlot+ 38 and PyMOL (http://www.pymol.org/pymol). The resulting interaction profiles were then compared with the receptor-ligand interactions obtained from docking using Autodock Vina.
MM/PB(GB)SA calculations
The binding free energies, for the protein−ligand complexes that passed the druglike and toxicity filters, were calculated using the MM/PB(GB)SA methods via the farPPI webserver. 45 GAFF2 and ff14SB force fields for the ligands and proteins, respectively, were used in combination to perform the calculations. The software was set to conduct PB3, PB4, GB1, GB2, GB5, and GB6 procedures simultaneously, just as reported by Rathod et al. 46
Molecular dynamics simulation
Molecular dynamics (MD) simulation of protein–ligand complexes was performed using the GROMACS simulation package via the WebGRO (available at https://simlab.uams.edu/) and the GROMOS96 43a1 force field was selected. The ligand topology files were generated using the GlycoBioChem PRODRG 2.5 server (http://davapc1.bioch.dundee.ac.uk/cgi-bin/prodrg). 47 Simple point charge (SPC) was selected as a solvent model (triclinic water box with size 50 Å × 75 Å × 70 Å) for the protein–ligand complex system. This system was neutralized by adding sodium or chlorine ions based on the total charges. For energy minimization of the system before MD, the steepest descent algorithm (5000 steps) was applied. The MD simulations were performed in the presence 0.15 M NaCl using the constant temperature (300 K) and pressure (1.0 bar). Approximate number of frames per simulation was 1000. These settings were selected to ensure that simulations were carried out under physiological conditions. The simulation time was set to 50 ns—the maximum duration provided by WebGRO, and the obtained trajectories of MD simulations were used for analysis of RMSD, root-mean-square fluctuation (RMSF), and hydrogen bonds (HBs).
Results and Discussion
Molecular docking simulation
In drug discovery, molecular docking is defined as a simulation that predicts the orientation of the ligand (drug) in the active site of a receptor (macromolecular target, eg, protein or enzyme). 48 Molecular docking programmes can calculate the ligand binding energy when it is bound with its respective binding site, creating a ligand-receptor complex. This facilitates the selection of the best ligand in virtual screening. 49 In this study, virtual screening of all 99 A. precatorius compounds was performed by molecular docking in the active site of SmPNP using AutoDock Vina. The binding energies (BEs) of docked compounds ranged from −5.0 kcal/mol to −10.4 kcal/mol. The co-crystallized ligand, inosine, had a BE of −7.8 kcal/mol. For subsequent steps, we selected only docked compounds that had BEs ranging from −8.0 to −10.4 kcal/mol, which is better when compared to the co-crystallized ligand. Seventy-four compounds passed the screen (see Table A.1 in the Appendix).
Validation of molecular docking accuracy
To validate the molecular docking protocol, the co-crystallized ligand, inosine, was docked into the binding pocket of SmPNP. The docked ligand was superimposed to calculate the RMSD value between the co-crystallized ligand and docked ligand (Figure 2b). In molecular docking validation, an RMSD value which is below 2.0 Å indicates that the employed docking protocol is good.50,51 In the present study, the calculated RMSD value was 0.55 Å indicating that the docking protocol was good. Further, we analysed the interacting amino acids to observe if the co-crystallized ligand and the docked ligand interacted with the active site amino acids in the same manner. Our results show that the co-crystallized ligand and the docked ligand interacted with the same amino acid residues and through the same bond types (Figure 2c). Overall, the validation results show that the docking protocol employed can be considered reliable for reproducing the binding pose of inosine in the X-ray crystal structure and, therefore, can be trusted for virtual screening.

X-ray crystal structure of SmPNP (PDB ID: 3FAZ), Chain A, in complex with inosine (a), Validation of molecular docking by superimposition of SmPNP co-crystallized ligand (blue) and the re-docked ligand (lime green) (b), 2-dimensional visualization of the re-docked reference ligand superimposed onto the co-crystallized complex using LigPlot+. The superimposed amino acid residues are shown in red circles. The green dotted lines indicate H-bonding between the ligand and amino acid residues, while residues in the half-circles represent the hydrophobic bond-forming residues (c).
Druglikeness analysis
The Lipinski et al’s RO5 has become a rule of thumb in determining the druglike properties of compounds. 39 According to Lipinski’s RO5, a drug like molecule should have a molecular weight not greater than 500 Da, hydrogen bond donors not greater than 5, hydrogen bond acceptors not greater than 10, and octanol-water partition coefficient (log P) not greater than 5. The significance of the Lipinski’s RO5 is that it helps to computationally predict if a compound is likely to have the physical and chemical properties to be orally bioavailable. In the current study, of the 74 compounds that passed the molecular docking virtual screening step, only 13 passed the Lipinski’s RO5. Table 1 shows the 13 compounds that passed the Lipinski’s RO5. Out of the 13 compounds that passed the Lipinski’s RO5, only 4 compounds passed the PAINS filter (Table 1). PAINS are compounds that often interact nonspecifically with multiple biological targets rather than selectively affecting the therapeutic target of interest. 42 Thus, these compounds usually give false-positive results in high-throughput screens and, therefore, are unsuitable to be lead compounds.42,52,53 The 4 compounds which have satisfied both the Lipinski’s RO5 and the PAINS filter are acceptable as druglike molecules.
Physicochemical properties of compounds that passed the Lipinski’s RO5.

Abbreviations: cLogP, partition coefficient; MW, molecular weight; H-A, hydrogen bond acceptor; H-D, hydrogen bond donor.
Toxicity risk assessment and drugscore
In the drug discovery process, toxic compounds often fail at several stages which is wastage of the invested costs, time, and human efforts.54-56 To obtain the best compounds which can be drugleads, the compounds which passed the RO5 and PAINS filter were computationally assessed for their toxicity properties.
The toxicity risk prediction results show that among the 4 tested compounds, only 1 compound, abrectorin, has a toxicity risk while the remaining 3 compounds (abrusogenin, hispidulin and cirsimaritin) are predicted to be safe. The results of the toxicity tests, with the drug scores of the respective compounds, are shown in Table 2. The drug score of a compound is calculated by combing contributions from Molecular weight, partition coefficient, solubility, and druglikeness and toxicity risk. The score is used to evaluate the potential of the drug candidate. The score ranges from 0 and 1, with 1 indicating a high possibility of a compound becoming a drug candidate and 0 indicating no possibility of being a drug candidate. Considering the results from molecular docking, druglikeness, and toxicity risk assessment, abrusogenin, hispidulin and, cirsimaritin. (Figure 3) may be exploited as promising drug candidates for the development of anti-schistosomiasis drugs.
The ttoxicity prediction profile and drug scores of the screened hits as calculated by OSIRIS Property Explorer.


2D-Structures of abrusogenin (a), hispidulin (b) and, cirsimaritin (c).
Visualization and analysis of receptor-ligand interactions
The 2D and 3D interactions of docked poses of top 3 compounds (abrusogenin, hispidulin and cirsimaritin) and amino acid residues within the SmPNP active sites are visualized in Figures 4 to 6, and summarized in Table 3. Abrusogenin forms 2 hydrogen bonds with Leu37 and Gly34 of 2.82 Å, and 2.76 Å, respectively, and hydrophobic bonds with 16 amino acids, viz, Cys33, Tyr90, His88, Asn117, Tyr202, Met221, Gly220, Val219, Val262, His259, Gly120, Ala119, Thr244, Leu263, Ala118, and Asn245 (Figure 4). The hydrogen bonds and hydrophobic interactions collectively yielded the binding energy of −9.8 kcal mol−1 as calculated by AutoDock Vina. As shown in Figure 5, hispidulin formed hydrogen bonds with Glu203 at a distance of 3.02 Å and Tyr90 at a distances 3.0 Å and 3.08 Å. In addition, the compound formed hydrophobic interactions with 12 amino acids Gly220, Met221, His259, His88, Gly34, Ala118, Asn245, Ala119, Tyr202, Val219, Gly120, and Asn117. The BE for the ligand is −8.6. Cirsimaritin, with the BE value of −8.2, forms 3 hydrogen bonds with Ser222 (3.06 Å) and Met221 (3.28 Å and 3.29 Å) and 9 hydrophobic bonds with Gly220, Ala118, Tyr202, His259, Ser35, Cys33, Gly34, Asn117, and Arg86 – Figure 6.

Hydrogen bonding and hydrophobic interactions of abrusogenin with SmPNP. 2D (a) and 3D (b) representations of its interactions and binding pose within the SmPNP binding site. Hydrogen bonds are shown by green dashed lines between the atoms involved and the donor–acceptor distance is given in Å, while hydrophobic contacts are shown by red arcs with spokes radiating towards the ligand atoms they contact.

Hydrogen bonding and hydrophobic interactions of hispidulin with SmPNP. 2D (a) and 3D (b) representations of its interactions and binding pose within the SmPNP binding site. Hydrogen bonds are shown by green dashed lines between the atoms involved and the donor–acceptor distance is given in Å, while hydrophobic contacts are shown by red arcs with spokes radiating towards the ligand atoms they contact.

Hydrogen bonding and hydrophobic interactions of cirsimaritin with SmPNP. 2D (a) and 3D (b) representations of its interactions and binding pose within the SmPNP binding site. Hydrogen bonds are shown by green dashed lines between the atoms involved and the donor–acceptor distance is given in Å, while hydrophobic contacts are shown by red arcs with spokes radiating towards the ligand atoms they contact.
Binding energies and protein-ligand interactions between the best ranked ligands and SmPNP as calculated by AutoDock Vina.

Bold, common interacting amino acids for all the 3 ligands.
The ability of the 3 top compounds to inhibit SmPNP apparently comes from a combination of hydrogen bonding and hydrophobic interactions.
Overall, the analysis of results receptor-ligand interactions show that both the hydrogen bonds and hydrophobic bods are important in maintaining the receptor-ligand complex. The common interacting amino acids for all the 3 ligands are Ala118, Asn117, Gly220, Gly34, His259, Met221 and Tyr202. This suggests that these residues are key in the binding of the ligand to the protein.
Re-validation of docking scores and receptor-ligand interactions
Before molecular dynamics, the binding scores and receptor-ligand interactions, of the 3 top-ranked compounds were further re-validated by BINDSURF Blind Docking Server. The binding energies calculated by BINDSURF were remarkably similar to and consistent with those generated by AutoDock Vina (Table 4).
Validation of binding energies and protein-ligand interactions between the best ranked ligands and SmPNP by as calculated by BINDSURF.

Bold, common interacting amino acids for all the 3 ligands.
In the case of abrusogenin, the AutoDock Vina generated interacting amino acids were the same as those generated by BINDSURF. For hispidulin, BINDSURF predicts that amino acid residue Gly120 interacts via H-bonding, while AutoDock Vina predicts that it is involved via hydrophobic interactions. Also, AutoDock Vina identified Asn117 to be involved in hydrophobic interaction with hispidulin. However, BINDSURF did not identify this amino acid as being in contact with the ligand. For cirsimaritin, docking using BINDSURF versus docking using AutoDock Vina generated the same interacting amino acids, except that His88 was only generated by BINDSURF.
Overall, BINDSURF adequately validated the molecular docking results obtained in this research.
Based on the validation results, we have confidence that the protein-ligand complexes generated by Autodock Vina are reliable and can be exploited further by molecular dynamics.
According to BINDSURF, the common interacting residues are Ala118, Gly220, His259, His88, Met221, and Tyr202.
MM/PB(GB)SA calculations
Since relying only on the docking scores does not satisfactorily predict the binding affinity between the protein and the respective ligands, we carried out free energy calculations using MM/PB(GB)SA to validate the docking energies of the protein–ligand complexes . The result for MM/PB(GB)SA using the PB3, PB4, GB1, GB2, GB5, and GB6 methods indicated that the complexes had good binding free energies(Table 5). Apart from GB6, all the procedures showed the same trend in ligand binding affinities as shown by molecular docking. That is, abrusogenin has a better affinity for the receptor followed by hispidulin and finally cirsimaritin. The GB6 method exhibited a different trend compared to other methods due to its unique approach to solvation modelling, which can influence the calculated binding affinities.
Calculated energy parameters for each of the complexes.

To sum up, the binding free energy results confirm that abrusogenin, hispidulin and cirsimaritin can bind efficiently and stably at the active site of SmPNP and, therefore, can be used as lead compounds.
Molecular dynamics
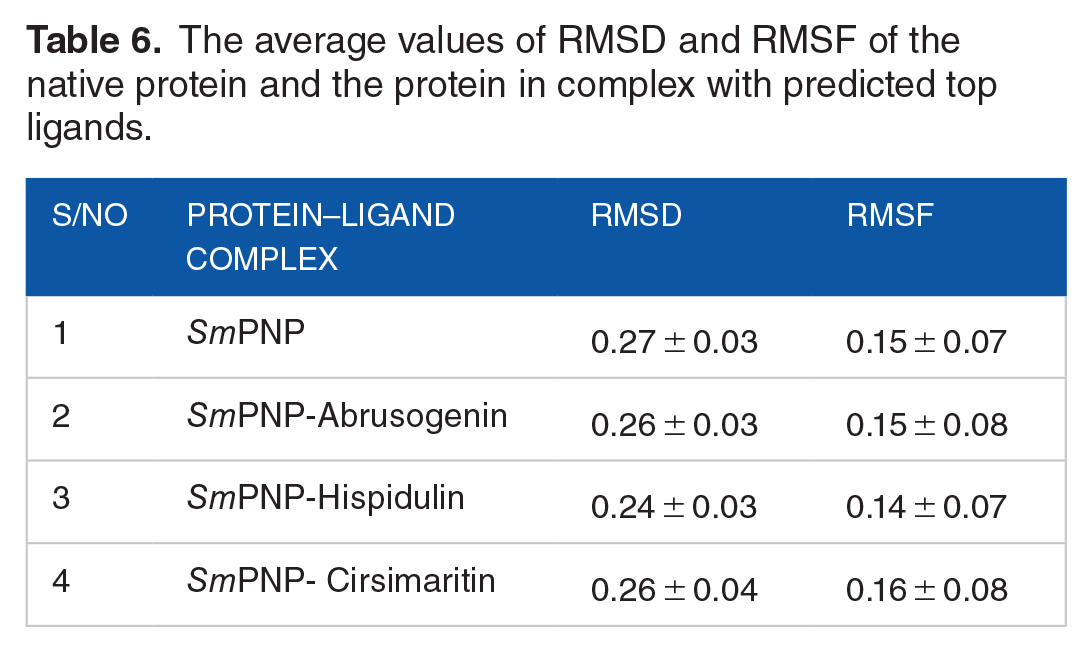
In computer assisted drug discovery approaches, MD simulations are commonly performed to evaluate the stability of the protein–ligand complexes. 57 We perfumed the MD simulations for abrusogenin, hispidulin and, cirsimaritin in the active site of SmPNP for 50 ns. For comparative purposes, the complexes were analysed alongside the unbound form of SmPNP. The RMSD, and RMSF results are summarized in Table 6.
The average values of RMSD and RMSF of the native protein and the protein in complex with predicted top ligands.
RMSD
For each complex, the RMSD of the protein backbone atoms were plotted against time to examine the variations in structural conformation. The deviations that occur during the simulation reflect the stability of the conformation. 58 Generally, a lower RMSD value implies fewer deviations in the system, whereas a higher RMSD value suggests that there are more structural fluctuations. 59 As shown in Figure 7, initially, all the complexes showed deviations till ~20 ns and then remained relatively stable till the end of the simulation. All the 3 complexes and the native protein showed a similar pattern of deviations. The observed minimal RMSD variations are acceptable as they show that the compounds are trying to stabilize their binding conformations at the binding pocket by giving flexibility to some of their chemical moieties, which forces the protein flexible loops to lose their original XYZ coordinates. 60 The overall average RMSD values were calculated to be 0.26931 nm (native SmPNP),0.26305 nm (SmPNP-Abrusogenin), 0.25952 nm (SmPNP-Abrectorin) and 0.24369 (SmPNP-Hispidulin). Generally, an RMSD value of less than 0.3 nm is acceptable for small globular proteins, 61 indicating that the complexes are stable.

RMSD analysis of the native protein (black) and its complexes with abrusogenin (red), hispidulin (green) and cirsimaritin (blue).
RMSF
The RMSF analysis represents the flexibility of each residue in the protein. RMSF was examined to analyse the changes in the behaviour of amino acid residues of SmPNP in the native state and on binding to the ligands (Figure 8). In all cases, the active pocket residues that interacted with the compounds remained highly stable during the simulation. The native protein, SmPNP, showed average fluctuations of 0.15075 nm while for the complexes SmPNP-Abrusogenin, SmPNP-Hispidulin and SmPNP- Cirsimaritin the RMSF values were calculated to be 0.15044 nm, 0.14451 nm and 0.15567 nm, respectively. The observed low RMSF values indicate less variation in the protein structure during the course of the simulation.

RMSF analysis of the native protein (black) and its complexes with abrusogenin (red), hispidulin (green) and cirsimaritin (blue).
H-bonds
The number of H- bonds present between interactions of the protein and ligand play an important role in the stability of that protein−ligand complex system. Thus, to examine the binding energy of the ligands with the target protein, the MD trajectories were analysed to interpret the extent of hydrogen bond formation during the entire course of simulation (Figure 9). All the complexes had formed H-bonds with the receptor protein throughout the simulation, indicating the stronger affinity of the ligands towards the target. This signifies the high intermolecular affinity of the docked compounds and receptor protein.

H-bonds analysis through the 50 ns simulation of SmPNP in complexes with abrusogenin (red), hispidulin (green) and cirsimaritin (blue).
Conclusion
We computationally identified 3 natural compounds (abrusogenin, hispidulin and cirsimaritin) as potential inhibitors of SmPNP. Apart from having high molecular docking scores, the compounds were analysed and predicted to have satisfactory physicochemical and toxicity properties. Further studies using binding free energy calculations and MD simulations, showed that the 3 compounds have good stability and dynamics within the SmPNP active site. In conclusion, our findings suggest that these proposed hits may be a promising starting point for the development of new schistosomicidal drugs based on natural products. However, further experimental research is needed to evaluate the efficacy of these compounds in vivo and in silico.
Footnotes
Appendix
Binding affinity (kca/mol) of the 99 A. precatorius docked into the active site of SmPNP.

| S/N | PubChem CID | Binding Affinity |
|---|---|---|
| 1 | 21629874 | −9.5 |
| 2 | 101679107 | −9.7 |
| 3 | 442106 | −7.1 |
| 4 | 3861164 | −7.1 |
| 5 | 5570 | −5.4 |
| 6 | 162943239 | −8.3 |
| 7 | 162943238 | −8.3 |
| 8 | 129685601 | −7.6 |
| 9 | 163015450 | −8.6 |
| 10 | 638072 | −6.2 |
| 11 | 1105 | −6.6 |
| 12 | 312822 | −8.6 |
| 13 | 5280934 | −6.1 |
| 14 | 163081991 | −7.8 |
| 15 | 162965363 | −7.8 |
| 16 | 158477 | −10.1 |
| 17 | 14982 | −8.7 |
| 18 | 44575701 | −10.1 |
| 19 | 162872928 | −9 |
| 20 | 9846221 | −8.7 |
| 21 | 102120183 | −8.3 |
| 22 | 102120184 | −9.5 |
| 23 | 16213698 | −9.9 |
| 24 | 5316321 | −10 |
| 25 | 12442897 | −9 |
| 26 | 14103656 | −8.4 |
| 27 | 78101175 | −9.7 |
| 28 | 73299 | −8.7 |
| 29 | 163039352 | −7.7 |
| 30 | 15628279 | −7.7 |
| 31 | 102120181 | −8.4 |
| 32 | 163042635 | −8.2 |
| 33 | 102120182 | −8.3 |
| 34 | 21594179 | −8.3 |
| 35 | 162967467 | −8.3 |
| 36 | 5315715 | −9 |
| 37 | 73810246 | −8.3 |
| 38 | 124061 | −8.7 |
| 39 | 162873536 | −8.3 |
| 40 | 85194335 | −8.3 |
| 41 | 101616458 | −9.2 |
| 42 | 10161657 | −9.3 |
| 43 | 6451058 | −9.8 |
| 44 | 6857683 | −9.7 |
| 45 | 44575936 | −8.9 |
| 46 | 441902 | −8.8 |
| 47 | 4483249 | −9.7 |
| 48 | 75049108 | −8.7 |
| 49 | 5257561 | −9.3 |
| 50 | 122544 | −7.8 |
| 51 | 86821 | −7.9 |
| 52 | 860 | −5.9 |
| 53 | 445639 | −5.2 |
| 54 | 5281116 | −5 |
| 55 | 965 | −5.7 |
| 56 | 8216 | −5.2 |
| 57 | 11005 | −5.3 |
| 58 | 985 | −5.7 |
| 59 | 160511 | −7.1 |
| 60 | 914 | −7.2 |
| 61 | 162895962 | −8.7 |
| 62 | 12526650 | −8.7 |
| 63 | 14488057 | −9.1 |
| 64 | 10317374 | −8.3 |
| 65 | 9907682 | −8.4 |
| 66 | 10070738 | −7.7 |
| 67 | 3009614 | −8.2 |
| 68 | 172847 | −8.2 |
| 69 | 162858192 | −8.3 |
| 70 | 85064920 | −8.3 |
| 71 | 56776396 | −7.7 |
| 72 | 44257521 | −8.2 |
| 73 | 9975772 | −8.2 |
| 74 | 102120185 | −8.3 |
| 75 | 163034502 | −9.5 |
| 76 | 163067810 | −8.8 |
| 77 | 44258566 | −8.7 |
| 78 | 163034501 | −9.5 |
| 79 | 11972325 | −8 |
| 80 | 11972327 | −8.1 |
| 81 | 5281628 | −8.6 |
| 82 | 90871502 | −8.5 |
| 83 | 101591983 | −8.6 |
| 84 | 44258417 | −8.5 |
| 85 | 14213594 | −8.5 |
| 86 | 14213596 | −8.6 |
| 87 | 101949505 | −8.1 |
| 88 | 74977939 | −8.2 |
| 89 | 162821301 | −9.7 |
| 90 | 44258341 | −9.7 |
| 91 | 188323 | −8.2 |
| 92 | 102158651 | −9.8 |
| 93 | 74977878 | −10.4 |
| 94 | 44257585 | −8.5 |
| 95 | 162948074 | −9.2 |
| 96 | 162821304 | −9.4 |
| 97 | 647833 | −6.8 |
| 98 | 114776 | −9.6 |
| 99 | 5280445 | −8.5 |
Funding:
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Declaration Of Conflicting Interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
RS conceived, and designed the study. All authors contributed to the implementation of the research, to the analysis of the results and to the writing of the manuscript. RS supervised the study. All authors have read and agreed to submit the final version of the manuscript.
Ethical Statement
Ethics approval was not required for this study.