
Abstract
Aim:
The Insilco study uses deep learning algorithms to predict the protein-coding pg m RNA sequences.
Material and methods:
The NCBI GEO DATA SET GSE218606’s GEO R tool discovered P.G’s outer membrane vesicles’ most differentially expressed mRNA. Genemania analyzed differentially expressed gene networks. Transcriptomics data were collected and labeled on P. gingivalis protein-coding mRNA sequence and pseudogene, lincRNA, and bidirectional promoter lincRNA. Orange, a machine learning tool, analyzed and predicted data after preprocessing. Naïve Bayes, neural networks, and gradient descent partition data into training and testing sets, yielding accurate results. Cross-validation, model accuracy, and ROC curve were evaluated after model validation.
Results:
Three models, Neural Networks, Naive Bayes, and Gradient Boosting, were evaluated using metrics like Area Under the Curve (AUC), Classification Accuracy (CA), F1 Score, Precision, Recall, and Specificity. Gradient Boosting achieved a balanced performance (AUC: 0.72, CA: 0.41, F1: 0.32) compared to Neural Networks (AUC: 0.721, CA: 0.391, F1: 0.314) and Naive Bayes (AUC: 0.701, CA: 0.172, F1: 0.114). While statistical tests revealed no significant differences between the models, Gradient Boosting exhibited a more balanced precision-recall relationship.
Conclusion:
In silico analysis using machine learning techniques successfully predicted protein-coding mRNA sequences within Porphyromonas gingivalis OMVs. Gradient Boosting outperformed other models (Neural Networks, Naive Bayes) by achieving a balanced performance across metrics like AUC, classification accuracy, and precision-recall, suggests its potential as a reliable tool for protein-coding mRNA prediction in P. gingivalis OMVs.
Introduction
Porphyromonas gingivalis (P. gingivalis) 1 is a Gram-negative, anaerobic bacterium keystone pathogen in periodontitis. P. gingivalis is also a prolific producer of outer membrane vesicles (OMVs), 2 which are small, membrane-bound structures that contain a variety of virulence factors. P. gingivalis OMVs have also been linked to several other diseases, including Heart, Stroke, Diabetes, Rheumatoid arthritis, and Systemic lupus erythematosus. The potential for P. gingivalis OMVs to cause systemic diseases is a growing area of research. As our understanding of these vesicles increases, new therapeutic strategies may be developed for preventing and treating periodontitis and other diseases.
They are typically 50 to 400 nm in size, composed of a single lipid bilayer derived from the bacterial outer membrane. They contain a variety of virulence factors, including fimbriae, gingipains, and lipopolysaccharide (LPS). 3 They can regulate neutrophils and macrophages and invade oral epithelial cells. They are involved in the pathogenesis of periodontitis and a number of other diseases. OMVs are produced by P. gingivalis as a mechanism for delivering virulence factors and other molecules to host cells.
Transcriptomics of mRNA transcripts includes protein-coding and non-coding transcripts that do not encode proteins. The prediction of protein-coding mRNA is a key step in many areas of biological research, including gene discovery, functional genomics, and drug discovery. Several methods can be used to predict protein-coding mRNA, 4 including Heuristic methods provide a quick and straightforward way to identify potential protein-coding regions based on predefined rules.
Deep learning, a subset of machine learning, has gained prominence for its ability to analyze large datasets and extract complex patterns. The process of developing a deep learning model involves several key steps. Initially, data preparation ensures the dataset is properly cleaned and formatted. Following this, the appropriate neural network architecture is selected based on the data’s nature (eg, convolutional neural networks for images). This study, however, utilizes a broader approach. In addition to deep learning, it employs Naïve Bayes and Gradient Boosting. Naïve Bayes, efficient for large datasets, analyzes individual sequence features to estimate class probabilities (protein-coding/non-coding). Gradient Boosting, a powerful ensemble method, builds on successive models to improve overall accuracy. These complementary approaches offer a robust framework for protein-coding mRNA prediction in P. gingivalis OMVs. Subsequently, the chosen model undergoes rigorous training and optimization. Algorithms like backpropagation adjust the model’s parameters iteratively until it achieves satisfactory performance. Through these steps, the model is refined to effectively tackle the specific problem at hand. By employing neural networks with multiple layers of interconnected nodes, deep learning models can autonomously learn features from input data. 5
The current study focuses on utilizing deep learning techniques to predict protein-coding mRNA sequences within P. gingivalis OMVs. This allows for the accurate identification of protein-coding regions within the transcriptome of P. gingivalis OMVs, thereby shedding light on the molecular mechanisms underlying pathogenesis. Furthermore, by incorporating structural and functional information from mRNA sequences, deep learning models can facilitate the discovery of potential therapeutic targets for periodontal disease and associated systemic conditions.6,7
Material and Methods
Using the GEO R tool on the NCBI GEO DATA SET GSE218606, the most differentially expressed mRNA was identified from outer membrane vesicles of P. gingivalis (Figures 1 and 2). The CYTOSCAPE algorithm was used to perform network analysis of differentially expressed genes. Differential gene expression data obtained was classified and labeled in to protein-coding mRNA sequence of P. gingivalis and protein-coding, nonprotein coding like pseudogene, lincRNA, bidirectional_promoter_lncRNA (Figure 3). The threshold value of FDR 0.05 was chosen as the cut-off criterion. Transcriptomics Data was Collected and involved gathering and labeling on protein-coding mRNA sequence of P. gingivalis and protein-coding, nonprotein coding like pseudogene, lincRNA, bidirectional promoter lncRNA.

Volcano plot of DEGs of upregulated and downregulated genes and with top differential gene expression includes-PTGS1, CXCL1, BDKRB2, CXCL8, SECTM1, SLC16A6, TFAP2C, and biologically involved in inflammatory response and chemokine signaling.
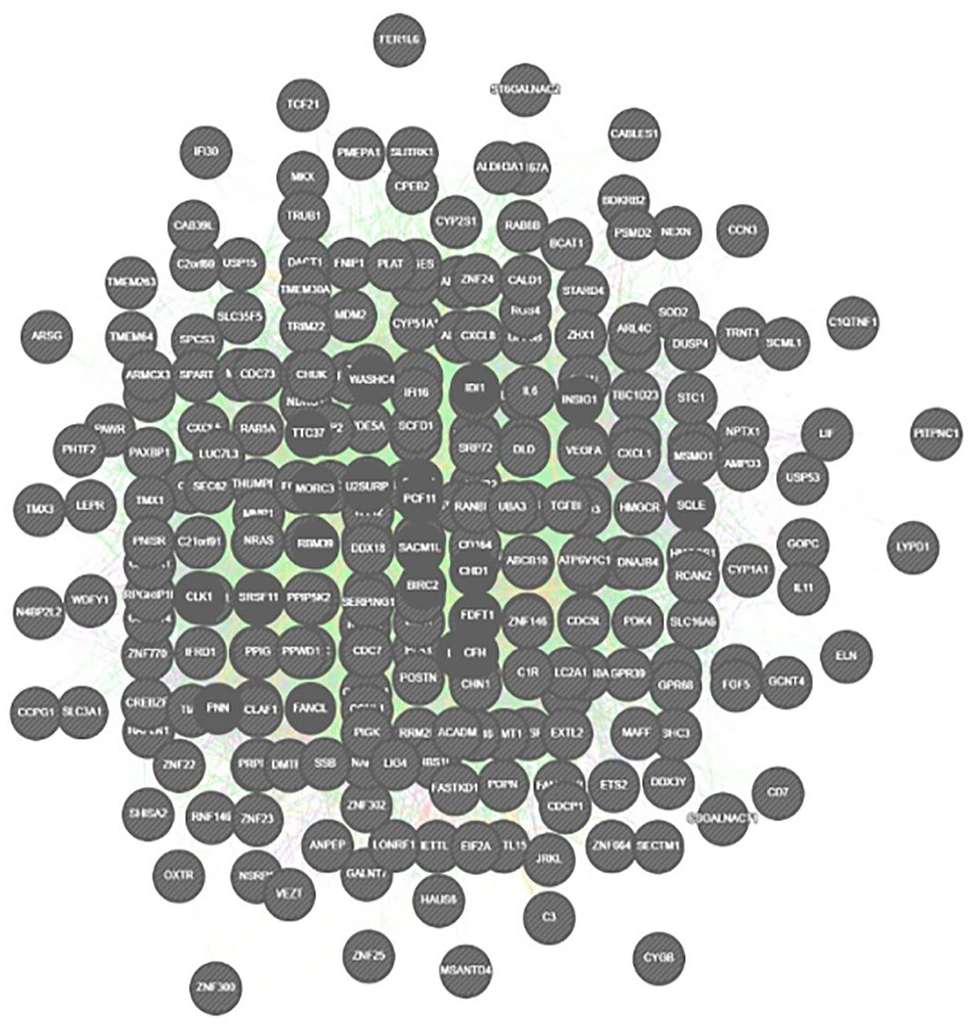
Interatomic hub genes of differential expressed genes.

Flowchart of machine learning model.
This research uses neural networks, Naïve Bayes, and gradient boosting to build a prediction model. Neural networks, Naïve Bayes, and gradient boosting are chosen for their effectiveness in different machine learning tasks. Neural networks are advantageous in handling high-dimensional data, while Naïve Bayes is a data mining technique that can be useful in solving various data-based problems. Gradient boosting, on the other hand, is a powerful machine learning technique that sequentially adds new models to the ensemble, improving the overall performance. Neural networks excel in pattern recognition, while Naïve Bayes is efficient for text classification. Gradient boosting, an ensemble method, combines weak learners for robust prediction models.
The network architecture consists of an input layer, hidden layers, and an output layer. It is initialized, initialized with random weights, and then used for forward propagation and backpropagation to generate predictions. The error between predictions and actual output is calculated, and the network is used to make predictions on new data. Gradient Boosting is a method that uses an ensemble of decision trees called weak learners, each built sequentially to correct previous errors. The final prediction is a weighted sum of these weak learners. The process involves initializing the model, fitting it to the training data, calculating the error, adjusting weights, building a new weak learner, and repeating until convergence. Naïve Bayes is a machine learning method based on Bayes’ theorem and feature independence. It uses probability distributions to estimate the likelihood of an instance belonging to a specific class. The process involves learning prior and conditional probabilities, and assigning the highest probability label.
Prior to analysis with machine learning models, the P. gingivalis OMV mRNA data underwent preprocessing. This initial step encompassed handling missing values (eg, through mean imputation or deletion), normalizing features for consistent interpretation, and potentially crafting novel features based on sequence motifs. Additionally, categorical data like gene names were likely transformed into numerical representations for optimal model comprehension. These preprocessing steps ensure the data is clean, standardized, and interpretable by the machine learning algorithms, ultimately improving the reliability of the analysis.
After preprocessing the data, Orange, a machine learning tool, was used for data analysis and predictive modeling activities. Machine learning splits involved using 80% of data for training and 20% for testing. The ideal split depends on the dataset size, problem complexity, and data availability. The split percentage affects model results, with smaller training sets potentially leading to bias and underfitting, and larger testing sets potentially causing overfitting. results were insensitive to percentage of selection. Hyperparameter tuning involved using 10 hidden layers, Adam optimizer, and ReLU activation for neural networks, while gradient boosting utilized 100 trees, a learning rate of 0.102, and trained on all instances. Cross-validation, model accuracy, and ROC curve were assessed. Once the model has been tested and validated, it can predict whether a given mRNA sequence codes for an inflammatory-inducing protein in P. gingivalis OMVs.
Recent advances in deep learning for bioinformatics suggest that these models could potentially consider not only the coding regions of the mRNA but also cis-regulatory regions such as promoters and untranslated regions (UTRs). Additionally, the structure of the mRNA and the corresponding protein could be crucial features for the model, as there is a strong correlation between protein and mRNA levels in multiple organisms.
Results
The presented Table 1 outlines the performance metrics for 3 distinct models—Neural Network, Naive Bayes, and Gradient Boosting—used to predict protein-coding mRNA sequences within Porphyromonas gingivalis outer membrane vesicles (OMVs). These metrics serve as evaluative tools to gage the effectiveness and accuracy of each model in discerning between positive and negative instances and to assess their overall performance. These metrics include AUC (Area Under the Curve), CA (Classification Accuracy), FI (F1 Score), Precision, Recall (Sensitivity), and Specificity. AUC measures the model’s ability to differentiate between classes, as it represents the area under the Receiver Operating Characteristic (ROC) curve. Higher AUC values signify better discrimination between positive and negative instances. CA represents the proportion of correctly classified instances among all instances, indicating the model’s overall accuracy. FI Score, a harmonic mean of precision and recall, offers a balanced assessment of the model’s performance, considering both false positives and false negatives. Precision measures the accuracy of positive predictions, while Recall quantifies the model’s ability to capture all actual positive instances. Specificity assesses the model’s ability to correctly identify negative instances.
Shows performance metrics for neural networks, naïve bayes, gradient boosting.

Beginning with the Neural Network model, it achieved a modest AUC of 0.721, indicating its discriminatory capacity, albeit with a relatively low classification accuracy (CA) of 0.391. Moreover, the F1 score was 0.314, suggesting a trade-off between these 2 metrics. Precision stood at 0.318, while recall was slightly higher at 0.391. However, the specificity, was notably high at 0.861, indicating robust performance in this aspect. Transitioning to the Naive Bayes model, it exhibited a slightly lower AUC of 0.701 compared to the Neural Network. However, its classification accuracy was considerably lower at 0.172, indicating poorer overall performance. The F1 score and precision were both significantly low at 0.114 and 0.418, respectively, indicating a substantial imbalance between precision and recall. Moreover, recall was found to be 0.172, suggesting that the model captures only a small portion of actual positive instances, while specificity remained relatively high at 0.902, indicating proficiency in identifying negative instances. Finally, the Gradient Boosting model demonstrated an AUC of 0.72, comparable to the Neural Network. However, it achieved a higher classification accuracy of 0.408, implying superior overall performance. The F1 score, serving as a measure of balance between precision and recall, was moderate at 0.322, indicating a reasonable trade-off between these 2 metrics. Precision and recall were both around 0.32 and 0.408, respectively, implying a fair balance between correctly identifying positive instances and minimizing false positives (Figure 4). Additionally, specificity was moderate at 0.843, indicating satisfactory performance in correctly identifying negative instances. These results collectively suggest that while all models exhibit some level of predictive accuracy, the Gradient Boosting model distinguishes itself for its superior overall performance and balanced performance across various metrics.

ROC curve of predictive accuracy of protein-coding and non-coding genes.
The Kruskal-Wallis H-test was used to compare the distributions of model metrics (AUC, Classification Accuracy, F1 Score, Precision, Recall, and Specificity) across Neural Networks, Naive Bayes, and Gradient Boosting. The results show that the H-statistic is 2.000 for all metrics, with corresponding P-values of .368. This indicates that there is no significant difference in the distributions of these metrics across the 3 types of models. statistically showed the P-values for all the metrics are higher than the usual significance level (eg, .05), which means that there is no statistically significant difference in the distributions of these metrics among the 3 models. This indicates that, based on the data, 1 model consistently performs better than the others across these metrics.
Discussion
Recently, there has been a growing interest in using machine learning methods to predict protein-coding mRNA. These methods can learn from large datasets of known protein sequences and their functions, and they can then be used to predict the importance of new proteins. P. gingivalis main virulence factors are fimbriae, capsule, outer membrane vesicles, LPS, toxic metabolites, and proteinases. Gingipains, “trypsin-like” cysteine proteinases, can degrade plasma, extracellular matrix, cytokines, and host cell surface proteins. P. gingivalis can concentrate and release OMVs with high virulence factors. Zhang et al 8 performed proteomics analysis of P. gingivalis OMVs and identified a total of 151 proteins, almost all of which were derived from the outer membrane or periplasm, and its protein composition is different from its parent bacteria.
P. gingivalis OMVs have been found to contribute to periodontitis through immunological mechanisms and bacterial virulence.3,9 Transcriptomics data used in this study were known for its involvement in periodontal disease. The data involved gathering and labeling protein-coding mRNA sequences.
In the case of transcriptomics data, the dependent and independent variables can vary depending on the specific analysis being performed. Generally, the independent variable or predictor variable would be the different RNA sequences, such as protein-coding mRNA sequences, pseudogenes, lincRNAs, or bidirectional promoter lncRNAs. These independent variables are used to predict or classify the dependent variable, which could be a specific biological outcome or phenotype. Regarding missing data, it is possible to have missing values in transcriptomics datasets. Handling missing data in transcriptomics data is removed. It is essential to carefully consider the potential impact of missing data and choose an appropriate method accordingly.
By labeling the mRNA sequences, they are assigned specific characteristics or attributes for use in machine learning algorithms. Additionally, the data also included protein-coding nonprotein coding sequences such as pseudogenes, lincRNAs (long intergenic noncoding RNAs), and bidirectional promoter lncRNAs. Pseudogenes are gene copies that have lost their protein-coding ability, while lincRNAs and bidirectional promoter lncRNAs are noncoding RNAs that have various functional roles within the cell.10,11 Machine learning algorithms can use transcriptomics data to identify patterns and associations between RNA sequences, potentially predicting new RNA sequences and identifying therapeutic targets for periodontal disease, based on identified patterns.
These protein-coding inflammatory-inducing mRNA markers are released from P. gingivalis OMVs and can induce inflammation and immune responses in host cells. This inflammation and immune response can lead to the destruction of periodontal tissues and the development of periodontitis. Generally, the mRNA data are large numbers, making it difficult to identify them manually. Suppose any future mutations occur or new proteins (Gingipains) or strains of P. gingivalis OMV are identified, and new mRNAs are identified. In that case, the task of detecting protein-coding mRNA gets tougher. Hence, having an AI model will help to reduce the workload of identifying the P. gingivalis protein-coding mRNA. The current study shows neural networks, naïve bayes, and gradient-boosting algorithms to be reliable in predicting the protein-coding mRNA. As new protein-coding mRNAs are identified, entering the data into the algorithms will help us automatically generate protein-coding mRNA.
Conclusion
The current study is one of its kinds in utilizing predictive models to detect protein-coding mRNA of P. gingivalis OMV. The 3 algorithms, neural networks, naïve bayes, and gradient boosting, tested in the study show better accuracy in predicting protein-coding mRNA. However, more algorithms should be tested to achieve reliable AI models.
Footnotes
Acknowledgements
None.
Author Contributions
P.K.Y.: Conceptualization and methodology, data collection and analysis, writing and editing, intellectual contributions and review. R.V.A.: Conceptualization and methodology, data collection and analysis, writing, editing and review. M.S.: Data collection, analysis and review. H.N.M.: Intellectual contributions and review. A.H.: Intellectual contributions and review.
Declaration of conflicting interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding:
The author(s) received no financial support for the research, authorship, and/or publication of this article.