
Abstract
New-onset refractory status epilepticus (NORSE) is a rare and devastating condition and the prognosis is often poor, with half to two-thirds of survivors experiencing drug-resistant epilepsy, residual cognitive impairment, or functional disability, and the mortality rate is 16% to 27% for adults. We describe a patient with cryptogenic NORSE and favorable recovery from drug-resistant super-refractory SE after the use of intravenous lidocaine. The patient experienced fever and presented with refractory generalized tonic-clonic seizures. The cause was not found by performing extensive examinations, including cell surface autoantibodies and rat brain immunohistochemistry evaluations. The refractory SE with unresponsiveness to multiple anti-epileptic and prolonged sedative medications, which are necessary for prolonged mechanical ventilation, were ameliorated by additive treatment with intravenous lidocaine initiating at 1 mg/kg/h and maintaining at 2 mg/kg/h for 40 days, which led to freedom from intravenous sedative medication and mechanical ventilation. The patient was able to return to school. Lidocaine may be an optional treatment for cryptogenic NORSE.
Introduction
New-onset refractory status epilepticus (NORSE) is a rare and devastating condition characterized by a highly drug-resistant refractory epileptic status and the presence of prodromal high fever of unknown origin. It occurs in previously healthy individuals without prodromal psychobehavioral or memory alterations before the onset of status epilepticus (SE). Approximately 20% of cases of refractory status epilepticus (SE) involve new onset refractory status epilepticus (NORSE). 1 In a retrospective review of 130 patients with NORSE, 52% had no known etiology despite extensive evaluation, and these patients were termed as having cryptogenic NORSE. 2 In 48% of the patients, the most frequently detected antibodies included anti-N-methyl-D-aspartate receptor (anti-NMDA), anti- voltage-gated potassium complex (VGKC), anti-leucine-rich glioma-inactivated 1 protein (LGI1) and, less commonly, anti-contactin-associated protein-2 (Caspr2). 2 Concerning NORSE-related treatments, standard anti-convulsant medications or sedative drugs are mostly administered to treat SE. No randomized controlled trials or prospective studies have been undertaken to guide immunotherapy. Based on expert opinion and case series, first-line immunotherapy agents including steroids, intravenous immunoglobulins, and plasmapheresis have been recommended to be administered as soon as possible, and tacrolimus, rituximab, cyclophosphamide, and anakinra in second-line treatment. 1 The prognosis of NORSE is often poor, with half to two-thirds of survivors experiencing drug-resistant epilepsy, residual cognitive impairment, or functional disability, including vegetative state, and the mortality rate is 16% to 27% for adults. 3 A useful treatment for cryptogenic NORSE is necessary because standard treatment, especially for adults, remains vague. We describe a patient with cryptogenic NORSE and favorable recovery from drug-resistant super-refractory SE after the use of intravenous lidocaine, suggesting that lidocaine may be an optional treatment for cryptogenic NORSE.
Case report
A 20-year-old healthy woman with no significant medical history or history of seizures presented with fever and diarrhea. Her family history was unremarkable. After 6 days, she presented with generalized tonic-clonic seizures and was admitted to the hospital. The Glasgow Coma Scale score was E4V3M6 without meningeal signs. Her body temperature was 38°C, but the physical examination results were otherwise unremarkable. No cranial nerve deficit or motor weakness was observed. The results of the routine laboratory examination were unremarkable. Cranial magnetic resonance imaging (MRI) showed some increased signals in the grey-white matter junction and at the cortical ribbon of both hemispheres (Figure 1). The cranial MR angiography findings were normal. Epileptiform activity such as spikes and waves, were predominantly evident in the frontotemporal area on the interictal electroencephalogram (Figure 2A). Generalized tonic-clonic seizures followed by twitching of the right side of her face and left hand persisted. Facio-branchio-dystonic seizures were absent. On examination, epileptiform activity was evident. On day 3, the seizures were frequent despite repeated intravenous diazepam administration, subsequently leading to hypoventilation, and our patient was admitted to the intensive care unit (ICU) whereupon 24-hour electroencephalography (EEG) monitoring was initiated. Cerebrospinal fluid (CSF) revealed a white blood cell count of 7/μL without an increased protein level (15 mg/dL). Polymerase chain reaction using viral and bacterial meningitis panels revealed negative cerebrospinal fluid results for Escherichia coli, Haemophilus influenzae, Listeria monocytogenes, Neisseria meningitidis, Streptococcus agalactiae, Streptococcus pneumoniae, cytomegalovirus, enterovirus, herpes simplex virus 1 and virus 2, human herpesvirus 6, human parechovirus, varicella zoster virus, Cryptococcus neoformans, and Cryptococcus gattii. Her cerebrospinal fluid (CSF) cultures for bacterial, tuberculosis, and fungal infection tested negative. The immunoglobulin G index increased (.75). She underwent three courses of methylprednisolone pulse therapy (1000 mg/day for 3 days), intravenous immunoglobulin (400 mg/kg/day for 5 days), plasmapheresis, and four courses of weekly rituximab (375 mg/m2) (Figure 3). Additionally, she received multiple anti-epileptic drugs, such as levetiracetam (maximum dose: 3000 mg/day), phenytoin (maximum dose: 400 mg/day), lacosamide (maximum dose: 300 mg/day), perampanel (maximum dose: 8 mg/day), valproic acid (maximum dose: 1600 mg/day), gabapentin (maximum dose: 1800 mg/day), and topiramate (maximum dose: 600 mg/day). However, she remained unresponsive to treatment and the generalized seizures continued to occur. The 24-hour EEG monitoring (Figure 2B) showed generalized spike and slow wave complexes approximately every 30 min despite the use of the following intravenous sedative drugs: propofol (maximum dose, 4 mg/kg/h); midazolam (maximum dose, .4 mg/kg/h); thiamylal (maximum dose, 5 mg/kg/h); and ketamine (1 mg/kg/h). When the dosage of three intravenous sedative drugs was tapered, convulsions occurred repeatedly; therefore, it was necessary to increase the dosage of the tapered sedative drugs. Under sedation, her body temperature was 38°C and she was comatose. Her pupils were 3 mm in diameter, equal, and reactive to light. The muscle tone decreased. Babinski’s sign was negative. Blood autoantibodies, including autonuclear antibodies and autoantibodies against GAD, MMP-3, CCP, ds-DNA, ss-DNA, SS-A/B, ANCA, GM1, and GQ1b, and anti-thyroid antibodies were all negative. Tumor markers, including AFP, CEA, CA19-9, CA125, CYFRA, NSE, and ProGRP, were negative. Enhanced computed tomography and MRI of the abdomen, enhanced computed tomography of the chest, upper and lower gastrointestinal endoscopy, echocardiography, and transesophageal echocardiography revealed no abnormal findings. At 20 days after admission, neuronal surface antibodies against antigens such as NMDA, LGI1, Caspr2, and AMPA were negative according to the cell-based assay. An immunohistochemical evaluation of the rat brain showed no reactivity with surface antigens. At 30 days after admission, repeated CSF showed that the interleukin-6 level (6.9 pg/mL), white blood cell count (1/μL), and protein level (15 mg/dL) did not increase. No oligoclonal bands of both serum and CSF were observed. These results support a diagnosis of cryptogenic NORSE. Axial cranial magnetic resonance imaging (MRI) taken 2 days post-admission. Both diffusion-weighted images (DWI) (left panel) and fluid-attenuated inversion recovery (FALIR) images (middle panel) showed some increased signals in the grey-white matter junction (white arrows) with decrease signal on the apparent diffusion coefficient (ADC) map, and increased signals at the cortical ribbon of both hemispheres. Panel A: Interictal electroencephalography (EEG) on day 1 post-admission. The patient displayed frequent twitches on the right side of her face and left thumb, followed by generalized convulsions. EEG revealed frequent sharp waves, mainly observed in bilateral frontotemporal areas. Panel B: 24-hour EEG monitoring (montages, monopolar; paper feed speed, 30 mm/sec; sensitivity, 200 μV/cm; high cut, 30 Hz; low cut, .3 Hz; notch filter, OFF) on day 71 post-admission. EEG showing generalized spike and slow wave complexes. Panel C: 24-hour EEG monitoring (montages, monopolar; paper feed speed, 30 mm/sec; sensitivity, 100 μV/cm; high cut, 30 Hz; low cut, .3 Hz; notch filter, OFF) on 96-hour post-lidocaine initiation. EEG showing no epileptiform activity. Clinical course of refractory convulsions, anti-seizure medications, and immunotherapies. LEV = levetiracetam; PER = perampanel; LCM = lacosamide; PB = phenobarbital; TPM = topiramate; GBP = Gabapentin; VPA = valproic acid; PHT = Phenytoin; IVMP = intravenous methylprednisolone; PE = plasma exchange; IVIg = intravenous immunoglobulin; RIT = Rituximab.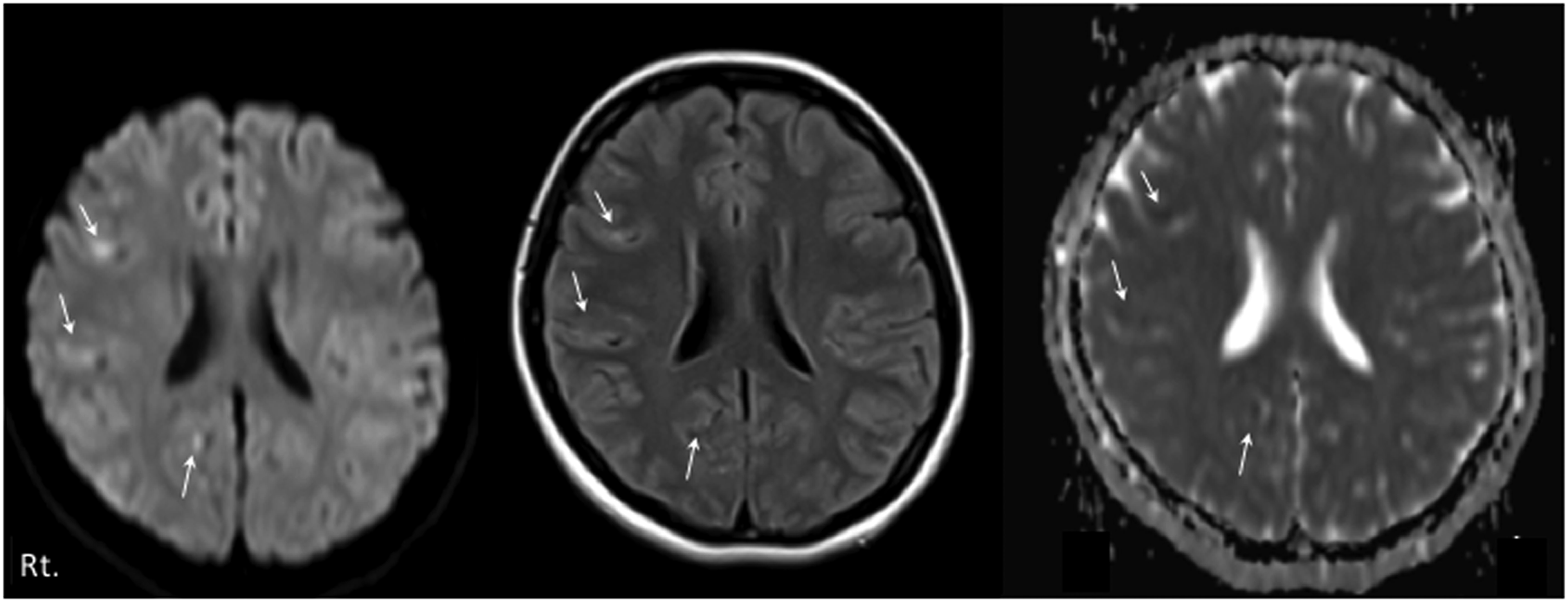

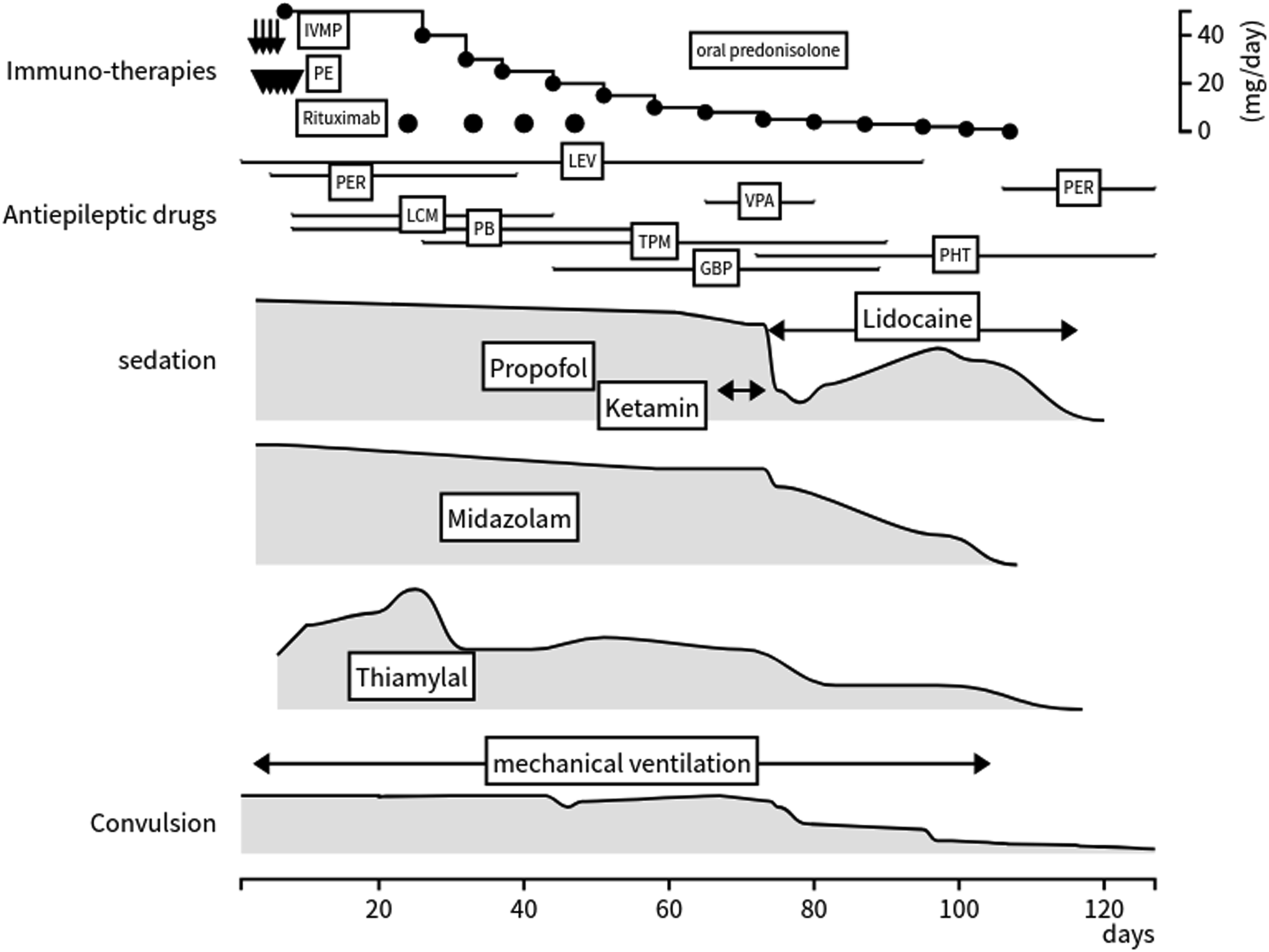
On day 74 after admission, continuous treatment with intravenous lidocaine was initiated at 1 mg/kg/h and maintained at 2 mg/kg/h for 40 days. At day 1 after lidocaine treatment was started, the magnitude and frequency of the convulsive seizures had markedly reduced. At 96 hours, sharp waves in the frontal and temporal regions (Figure 2C), which had occasionally been observed prior to lidocaine initiation on continuous EEG monitoring, were absent. During lidocaine infusion, a temporary increase in the propofol dosage was required while carefully tapering both midazolam and thiamylal. We administered anti-epileptic drugs such as gabapentin, and she was newly started on phenytoin and restarted on perampanel. Additionally, drug-induced febrile neutropenic syndrome unrelated to the lidocaine, which was prescribed to control the infection, co-occurred. The magnitude and frequency of her seizures gradually diminished and all sedative drugs were discontinued once her seizures had stopped (at 32 days post-lidocaine initiation). Cranial follow-up MRI, which was performed once a week for 120 days during ICU admission, showed that the increased signals in the grey-white matter junction and at the cortical ribbon of both hemispheres had disappeared. She had no memory impairment and retained all her premorbid memories. She was transferred to another rehabilitation hospital on day 230 after admission because of mild weakness and spasticity of the lower limbs. At 5 months after admission, the patient was able to return to school.
Discussion
Our patient had new-onset refractory SE. The cause was not found by performing extensive examinations, including cell surface autoantibodies and rat brain immunohistochemistry evaluations, thus supporting the diagnosis of cryptogenic NORSE. The magnitude and frequency of refractory SE with unresponsiveness to multiple anti-epileptic and prolonged sedative medications, which are necessary for prolonged mechanical ventilation, were ameliorated by additive treatment with intravenous lidocaine, which led to freedom from intravenous sedative medication and mechanical ventilation and resulted in sufficient recovery. The combinations of lidocaine with levetiracetam and MgSO4, 4 high-dose topiramate, 5 and levetiracetam and sodium valproate 6 for cryptogenic refractory SE for children 5 to 15 years of age have been reported. One patient with intractable epilepsy showed a favorable and prolonged clinical response over the course of 10 years after combination therapy with lidocaine taper and subcutaneous lidocaine infusion in addition to anti-epileptic drugs. 7
Lidocaine, a local anesthetic drug and anti-arrhythmic drug (class IB), has membrane-stabilizing effects and central local anesthetic action via inhibitory pathway fibers involved in direct cortical stimulation. The mechanism of lidocaine administration in refractory SE remains unclear. Sodium channel-based anti-epileptic drugs have a diphenyl motif, and the binding site blocks sodium ion transport and inactivates sodium channels in the central nervous system. 8 Lidocaine does not contain diphenyl structures, but it has one aromatic group with an amine chain. 9 Electrophysiologic studies involving cell biology have shown that carbamazepine and lidocaine bind to overlapping sites with the common aromatic motif and produce additive inhibitory effects on Na+ currents in the channel pores. 9 The benefits of lidocaine for refractory seizures may be additive in the presence of anti-epileptic drugs that inactivate the sodium channel.
There is currently a greater level of evidence and more extensive recommendations concerning immune-based therapies for adult patients with SE than for the use of lidocaine in such patients. 10 The seizure response rate to lidocaine administration in a population with SE has not previously been reported. Several pediatric studies have reported on the usefulness of lidocaine in the treatment of refractory SE. One survey of 194 neonatal ICUs at university hospitals in Japan showed that seizure frequency rates had reduced in an estimated 81.3% of 194 patients who had received lidocaine. 11 However, data regarding adults are limited in case reports, which have shown various efficacy rates; therefore, the efficacy of lidocaine and its potentially serious adverse effects for adults have not been well-defined. Refractory SE could not be controlled in a 49-year-old; 12 however, it was successfully controlled in a 15-year-old 6 and 23-year-old. 13 Italian guidelines for adults recommend lidocaine (grade C, evidence level 4) as an alternative treatment for refractory SE when other therapeutic options have failed. 14 A review of the treatment of children and adults with super-refractory SE has provided insufficient evidence to support the effectiveness of lidocaine. 15
The proposed neuroinflammation-mediated epileptogenesis remains largely unknown, 16 and the use of immune-therapies has not resulted in compelling evidence. The interleukin-1 receptor antagonist anakinra 17 and interleukin-6 receptor antagonist tocilizumab 18 showed potential efficacy for patients with cryptogenic NORSE. Increased levels of cytokines and chemokines have been reported.17-19 Our patient had a fulminant clinical cause despite intensive immune therapies, including repeated rituximab, which is a monoclonal antibody that binds to the differentiation antigen CD20 present on the surface of human B lymphocytes. However, lidocaine administration can lead to subsequent favorable effects when immunotherapy treatment precedes lidocaine administration.
In our patient with cryptogenic super-refractory SE, the use of lidocaine led to a favorable outcome. Lidocaine may be considered a treatment option owing to its potential effect on cryptogenic super-refractory SE. Such cases are rare among adults; therefore, confirmation of our treatment is required in future studies.
Footnotes
Acknowledgements
We are extremely grateful to all clinicians and other medical stuffs who did clinical practice for the present patient.
Author contributions
M Sugata, Y Uchihara, D Shimada, K Atagi, M Nakamura, M Hara, and M Kawahara contributed to acquisition of data. M Sugata, H Kataoka and M Hara contributed to analysis and interpretation of data. M Sugata H Kataoka and S Kazuma contributed to drafting and critical revision of part of the submitted materials.
Ethical Statement
Declaration of conflicting interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding:
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Evaluating cell surface autoantibodies and rat brain immunohistochemistry in the present case is partly supported by MHLW Grant Number 22HA1003 and JSPS KAKENHI (grant No. JP20K07875) (Makoto Hara).