
Abstract
IRF2BPL gene variants have recently been associated to developmental disability and epilepsy in children and movement disorders in adults. So far, only few cases have been reported; here we present four novel cases identified by exome sequencing, while investigating developmental delay, adult-onset cerebellar ataxia or regression.
Keywords
Introduction
In 2018, a novel syndrome associated with pathogenic variants in IRF2BPL gene was delineated, which includes developmental delay, regression, epilepsy, movement disorders, ataxia, pyramidal signs, vertical gaze palsy, and dysmorphic features.1,2 This condition was named “neurodevelopmental disorder with regression, abnormal movements, loss of speech, and seizures” – NEDAMSS (MIM #618088). It has been proposed that nonsense variants cause regression, movement disorders and severe developmental delay, whereas missense variants were associated with developmental delay and seizures. 1 We report on four new patients with novel variants of this gene.
Case 1
An 18-month-old girl presented with global developmental delay. She is the second child of a non-consanguineous couple and there is no reported family history of malformations or developmental delay.
Pregnancy was complicated by gestational diabetes after the 7th month. The child was born at 39 weeks and 1 day by c-section due to pelvic presentation. Birth weight was 3860g (p90-p97), and length 51 cm (p90-97). She was diagnosed with hip dysplasia during the neonatal period, and treated non-surgically with Pavlik harness and body cast until 6 months old. After cast removal, hypotonia became evident, with referral for neurological evaluation at the age of 8 months.
She was able to stand without support at the age of 13 months and to walk without support at 2 years. She spoke simple words at the age of 2 years and was later diagnosed with speech apraxia by a speech therapist. She never presented with epilepsy or developmental regression.
Clinical exam revealed subtle dysmorphic features such as ectropion of lower eyelids, sparse eyebrows, posteriorly rotated ears, prominent fingertip pads, long fingers, and pectus carinatum. Weight, height, and head circumference were within the normal range at the ages of 18 months and 4 years 3 months. (Figure 1). Patient 1: Note sparse eyebrows, ectropion of lower palpebra.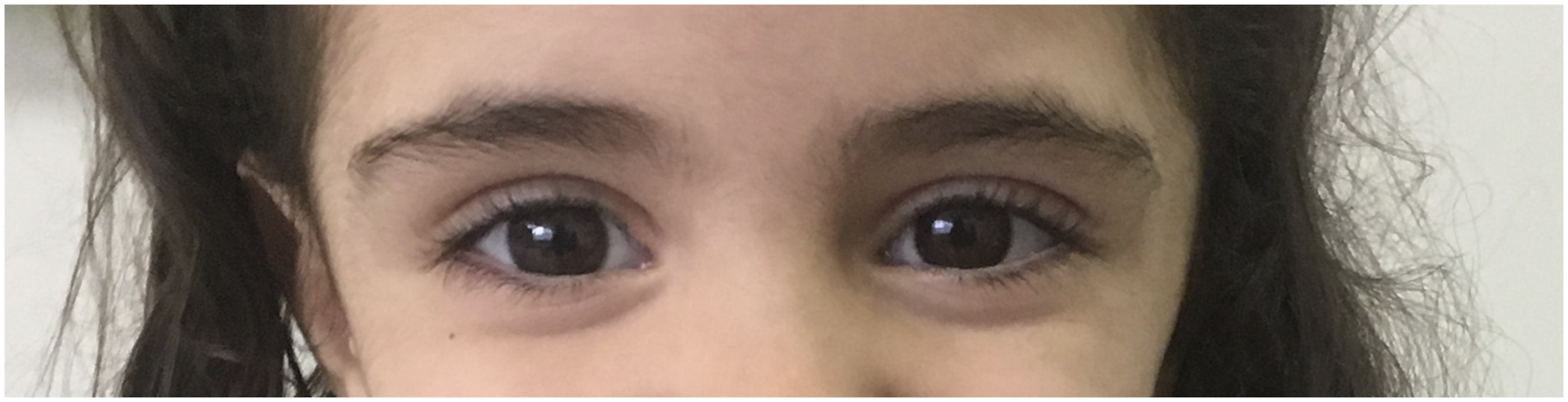
Clinical investigation included G-band karyotype, array-CGH, electroencephalogram, abdominal ultrasound, echocardiogram, ophthalmological evaluation, auditory evoked potentials test, brain MRI; no abnormalities were found. Exome sequencing revealed a heterozygous variant classified as pathogenic on IRF2BPL – c.2102del; p.Asn701ThrfsTer66 (NM_024496.4). Moreover, a probably pathogenic variant was identified in another gene: HCN1 – c.1246 C>T; p.Gln416Ter (NM_021072.4). Both variants were de novo.
Case 2
A 47-year-old woman presented with cerebellar ataxia and parkinsonism. Although she had gait imbalance since the age of 20, her ability to do daily activities and self-care were never been impaired before. She was prescribed levodopa at the age of 52 years, with mild response in bradykinesia.
At the age of 55, she presented with severe speech impairment and disrupted sleep pattern. Benzodiazepine was prescribed for insomnia, but induced hallucinations. Polysomnography revealed diminished REM sleep, shortening of N3 stage of sleep, and frequent episodes of waking up during the night, but did not confirm a REM sleep disorder. Currently, at the age of 57 years, she needs aid for all daily living activities and is wheelchair bound.
Information about developmental milestones was not available. However, her family denies any cognitive or motor impairments during childhood. She graduated from university and used to work as a school supervisor.
Family history was negative for cerebellar ataxia or parkinsonism. She is the only child of a non-consanguineous couple, and unfortunately no information was available for the paternal family. Her mother presented with mild intellectual disability, was diagnosed with hydrocephalus at the age of 13 years and died at the age of 52 years due to hypertension.
The first clinical evaluation of patient 2 by a medical genetics’ specialist was at the age of 48 years. Her clinical exam revealed preserved strength, upper vertical gaze palsy, rigidity, bradykinesia, incoordination, hyperreflexia in upper limbs and areflexia in lower limbs, Babinski sign on the right foot, and mild dysarthria. No dysmorphic features were observed.
Memory and cognitive abilities were never formally evaluated. She performed 27 out of 30 points in the mini-mental test. She didn’t report memory loss or difficulties to manage daily activities within the limits of her motor disabilities.
Her clinical investigation included molecular analysis of SCA1, SCA2, SCA3, SCA6, SCA7, SCA10, SCA12, SCA17, DRPLA; dosing of chitotriosidase, vitamin E, copper, ceruloplasmin; beta-glucuronidase activity, hexosaminidase activity, plasma amino acid chromatography, Fillipin test, urinary organic acids; all tests revealed normal results. Electroneuromyography at the age of 50 years did not disclose any abnormalities, nor did brain MRI.
Exome sequencing revealed an heterozygous probably pathogenic variant on IRF2BPL – c.590del; p.Asn197ThrfsTer15 (NM_024496.4). Parents were not available for testing.
Case 3
A 5-year-old boy presented with global developmental delay. He was able to walk without support by the age of 14 months. Although simple words were spoken at the age of 1 year, he only used simple sentences after the age of 3 years.
He was born by c-section after an uneventful pregnancy, post-term (42 weeks), weighting 3700 g (p50), length 51 cm (p50), no intercurrences during neonatal period.
Several upper airways infections and constipation were observed until the age of 3 years. Clinical evaluation revealed hypotonia, upslanted palpebral fissures, epicanthus inversus over the left eye, hypertelorism, posteriorly rotated ears, overfolded helix, anteverted nares, synophrys, high, narrow palate, prominent fingertip pads, slender fingers, and joint laxity (Figures 2 and 3). Currently he is 10 years old and shows impaired social interactions. No seizures or regression were observed so far. Patient 3 at 7 years of age: Upslanted palpebral fissures, epicanthus inversus over the left eye, overfolded helix, synophrys. Patient 3 at 7 years: Upslanted palpebral fissures, epicanthus inversus over the left eye, overfolded helix, synophrys.

He is the first child of a non-consanguineous couple with no family history for malformations or developmental delay.
Clinical investigation included G-band karyotype, array-CGH, electroencephalogram, abdominal ultrasound, echocardiogram, brain MRI; all were within normal range. Exome sequencing revealed an heterozygous de novo variant classified as pathogenic on IRF2BL – c.1099 G>T; p.Glu367Ter (NM_024496).
Case 4
A 13-year-old boy presented with progressive walking impairment, gait instability and cognitive decline. The family also reports that behavioral problems became evident and school performance declined over the years. He presented tonic-clonic seizures at the age of 16 years and was started on oxcarbazepine. At the age of 17 years, he presented dysphagia and dysarthria.
He was born by c-section, at term, after an uneventful pregnancy. His birth weight was 3000 g, no other information from birth was available. The family reports motor development within the normal range and no speech issues during infancy. However, until the age of 3 years, feeding was difficult due to chewing problems.
He is the only child of a non-consanguineous couple with no family history of genetic or neurological diseases.
Clinical exam revealed ataxic gait, impairment of strength in lower limbs, and no dysmorphic features were observed. Brain and spine MRI showed no abnormalities. Electroneuromyography revealed proximal myopathy; muscular enzymes were the within normal range. Exome sequencing disclosed a pathogenic variant on IRF2BPL – c.474_504del, p.Ala161SerfsTer8 (NM_024496.4). We could not evaluate this variant in his parents.
Discussion
IRF2PBL variants have been associated with neurological symptoms from childhood to adulthood. Most patients reported so far had frameshift or nonsense variants. IRF2BPL is an intronless gene, which translates to a sequence of 796 aminoacids. Rampazzo et al. 3 suggested that this is a housekeeping gene, as it lacks a TATA-box upstream the origin of transcription.
First studies about IRF2BPL suggested that its function was regulation of female reproduction. 4 Recently, Marcogliese et al. 5 identified that overexpression of IRF2BPL downregulates expression of Wnt. Therefore, when IRF2BPL loses its function, Wnt is overexpressed and leads to neuronal degeneration. The majority of intronless genes encrypt receptors, regulatory and signaling molecules. Also, they escape nonsense mediated decay. 1
We report on three new frameshift variants and a new nonsense variant that are predicted to insert a premature stop codon. Most variants reported so far are nonsense or frameshift (Figure 4). Even though pathogenic variants have been reported throughout the gene, the majority are localized in the polyglutamine tract or before the first PEST sequence of the gene.1,2,6-13 A PEST sequence is a proline, glutamate, serine, threonine rich region that serves as signal for rapid proteolytic degradation.
14
There are 3 regions predicted to be PEST motifis in this gene (positions 201-213, 331-342 and 545-559). Interestingly, in all four patients (one reported here) who presented symptoms in adult life,6,12,15,16 the variants were localized in this region. Whether this region represents a cluster for adult-life presentations of the disease is still not known.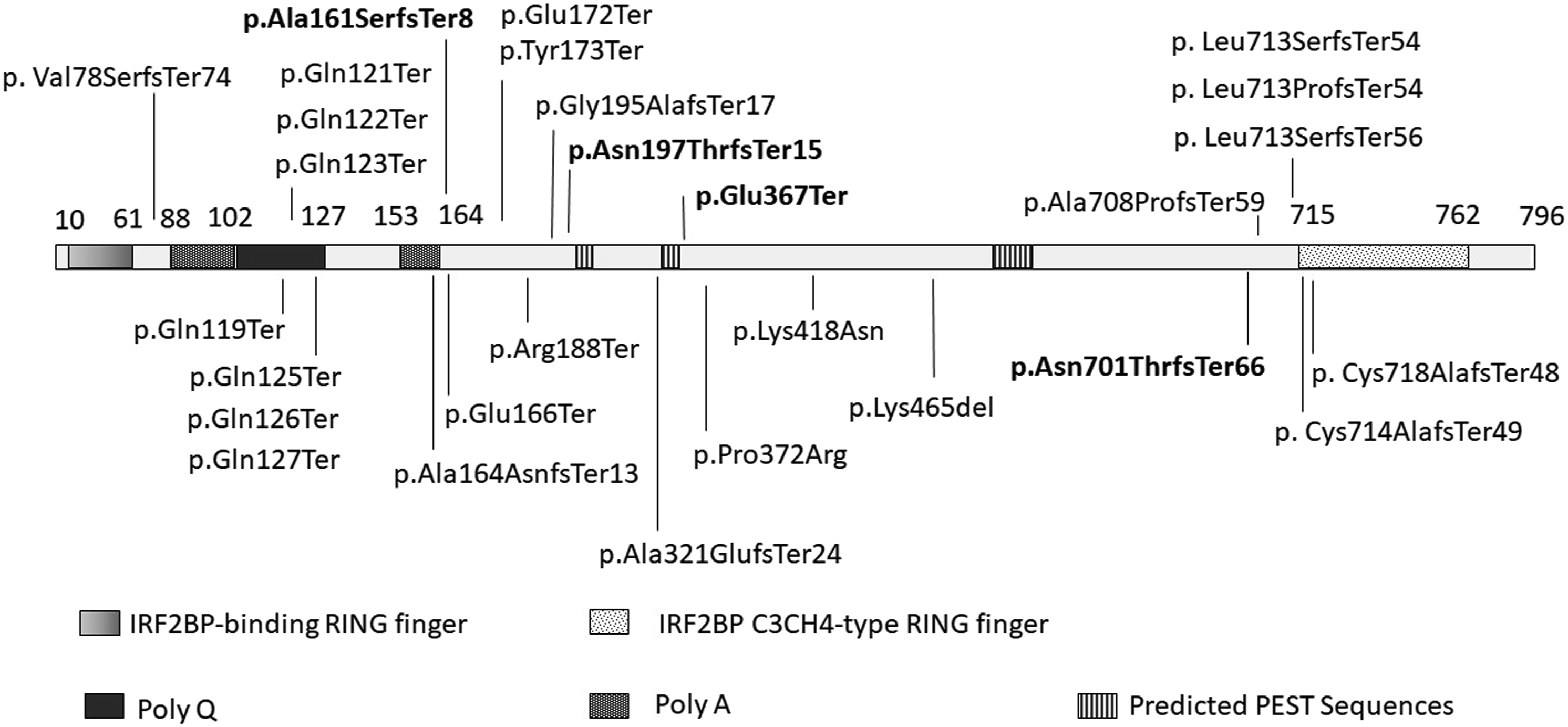
In case 1 two pathogenic variants were identified, one in IRFBPL2 gene and another on HCN1 gene. HCN1 gene has been associated to heat sensitive seizures, epileptic encephalopathy and generalized seizures. Some patients also present intellectual disability, global development delay, attention-deficit hyperactivity disorder, behavioral disturbances. 23 This gene is part of a family of hyperpolarization-activated cyclic nucleotide-gated channels which are expressed in the brain and the heart. It has a role in controlling spontaneous activity of cardiac rhythm and neuronal excitability among other functions. 24 Nava et al. 25 reported 6 patients with missense variants in this gene who presented epileptic encephalopathy or a Dravet-like syndrome. Interestingly, the variants identified in this study had a gain-of-function or dominant negative effect. Bonzanni et al. 26 and DiFrancesco et al. 27 reported missense variants in patients with epilepsy who did not present developmental delay or epileptic encephalopathy. Epilepsy is usually secondary to loss of inhibitory mechanisms. Nevertheless, HCN1-epilepsy has been associated to both loss of function and gain of function variants. 23
As the HCN1 nonsense variant was identified in a patient with developmental delay, at this moment we cannot define if her symptoms are secondary to IRF2BPL variant or the HCN1 variant. She did not present seizures and electroencephalogram did not reveal any epileptic activity up to the age of 4 years. The laboratory classified the HCN1 variant as likely pathogenic based on ACMG criteria (PVS1, PS2, PM2, BP5). Whether this variant represents a new variant with incomplete penetrance, age-dependent penetrance or if its only presentation is related to developmental delay is unknown at this moment.
The second patient is the oldest person reported so far. Interestingly, she presented symptoms in adult life, and unlike the other adult patients reported, she does not seem to present with severe cognitive decline, which may be observed in the following years, as regression is an expected symptom. She has already lost many of her motor skills. Symptoms of sleep disorder were suggestive of REM sleep behavior disorder. This has been associated with α-synucleionopathy neurodegenerative diseases, neurological disorders, or antidepressant medications. 28 Diagnosis requires video polysomnography, which was not performed in our case. In the cases published so far, manifestations of sleep disorders have not been described in detail. As loss of IRF2BPL leads to overexpression of Wnt 5 and this pathway has been associated with insomnia, 29 it is interesting to further investigate sleep patterns in patients with NEDAMSS.
The third patient seems to have a very mild presentation of IRF2BPL impairment. Even though speech delay was most evident when he was very young, his intellectual abilities are evolving. Nowadays his major impairment is related to social interaction. Even though regression has not been observed in cases 3 and 4, it may still happen.
Dysmorphic features were observed in 7 out of 11 patients described by Mau-Them et al. 2 However the authors did not provide a full description of them. Shelkowitz et al. 7 mentioned a few dysmorphic features of which hypertelorism was also observed on patient 3. At this point, it is not possible to define if dysmorphic features are an important manifestation of this condition.
Epilepsy is a main feature of NEDAMSS and has been reported in more than half of the cases.1,2,7-10,13,15-19 Some variants were even identified while screening for monogenic diseases among epilepsy patients13,16,18 and, it has been proposed that this gene should be included in epilepsy panels. However, in none of the cases reported here, seizures were a manifestation. Therefore, NADAMSS should be considered in the differential diagnosis of neurodevelopmental disorders, even in cases that seem mildly affected.
IRF2BPL should also be considered in the differential diagnosis of adult patients with parkinsonism, ataxia, and cognitive decline. Recently, this gene has been recognized as a dystonia associated gene with a generalized dystonia phenotype. 30 It is important to carefully document all clinical, laboratory and pathological findings, as this will help increase our knowledge about this condition.
Footnotes
Author Contributions
All authors participated equally in the acquisition, analysis and interpretation of data. Maria Angelica de F. D. de Lima drafted the manuscript and all authors reviewed this manuscript. All authors approved this version.
Declaration of conflicting interests:
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding:
The authors received no financial support for the research, authorship, and/or publication of this article.
Ethics statement:
Hospital Universitário Gaffrée e Guinle Ethical Review Board approved the procedures here (CAAE: 31416920.7.0000.5258; date: June 30th, 2020). Written informed consent was obtained from patient 2 and from parents of patients 1, 3 and 4 for this research as well as for publication of patient data and images. Consent was registered via consent statement form previously approved by Gaffrée and Guinle University Hospital Ethical Review Board.
Data availability:
All data used to write up this manuscript is available for review upon a reasonable request.