
Abstract
Since Alois Alzheimer described the pathology of Alzheimer’s disease in 1907, an increasing number of studies have attempted to discover its causes and possible ways to treat it. For decades, research has focused on neuronal degeneration and the disruption to the neural circuits that occurs during disease progression, undervaluing in some extent the alterations to glial cells even though these alterations were described in the very first studies of this disease. In recent years, it has been recognized that different families of neuroglia are not merely support cells for neurons but rather key and active elements in the physiology and pathology of the nervous system. Alterations to different types of neuroglia (especially astroglia and microglia but also mature oligodendroglia and oligodendroglial progenitors) have been identified in the initial neuropathological changes that lead to dementia, suggesting that they may represent therapeutic targets to prevent neurodegeneration. In this review, based on our own studies and on the relevant scientific literature, we argue that a careful and in-depth study of glial cells will be fundamental to understanding the origin and progression of Alzheimer’s disease. In addition, we analyze the main issues regarding the neuroprotective and neurotoxic role of neuroglial changes, reactions and/or involutions in both humans with Alzheimer’s disease and in experimental models of this condition.
Keywords
Introduction
Alzheimer’s disease (AD) is a neurodegenerative syndrome that leads to dementia and was first described more than a century ago by Alois Alzheimer. 1 A few years ago, AD was described as an “epidemic of the 21st century”, both due to the large number of cases expected (more than 20 million people by 2020) and the enormous economic and social burden it places on countries around the globe. 2 For many decades, researchers from different disciplines around the world have been attempting to find the causes of this neurodegenerative process and to find a treatment to prevent or treat it once dementia has set in.
Since the original description of the disease, research into AD has mainly focused on assessing the alterations to the neurons and circuits that might provoke the cognitive decline associated with dementia (the “neuron-centric” theory of AD).3-7 Over the years, different techniques (neuropathological, biochemical, biomolecular, genetic, etc.) have been used to study neurons in normal and pathological situations, both in “healthy” and “Alzheimer” individuals, as well as in experimental models of AD.8-18 In most human studies and in many pathogenic models of AD, was considered that glial cells also “suffer concomitant alterations”, and these alterations are thought to aggravate neurodegeneration,19-32 representing key changes in the pathogenic cascades that provoke neurodegeneration/dementia. However, the specific study of glial cells in AD begins many decades after (except for exceptions that are later mentioned). It was necessary to develop new technologies to study the types and subtypes of glial cells and their functions, as well as to understand and accept that these cells were intimately involved in the functions of neurons and neuronal circuits, to focus on them the investigation of neurodegenerative disorders.22-31 As such, in recent years, astrogliosis and microgliosis, the main drivers of neuroinflammation,24,25,27-31,33-39 as well as changes in oligodendroglial cells, have been integrated into all the pathogenic theories of AD.33-36,40 In fact, we can even consider that “neuroglial” theories of AD now exist.
The importance of glia in AD was recognized long ago by the Spanish neuroscientist Nicolas Achúcarro, a collaborator of both Alois Alzheimer and Santiago Ramón y Cajal. In Achúcarro’s studies (1910-1925), he pointed out how astroglia and microglial cells undergo important changes during human neurodegeneration, indicating that they should not be underestimated.3,19-21,26,41 However, there was a lack of technologies capable of differentiating subtypes of neuroglial cells and their functions to analyze their real involvement in the disease. In more recent decades, new research supported by new technologies (histochemical, biochemical, genic, and so on), significantly increased our understanding of glial cells (23-36, 40). Accordingly, glial cells are no longer merely considered support cells for neurons and neuronal circuits; instead, they are thought to function very actively in maintaining the morphology and correct functioning of the nervous system as well as in the adaptive responses on which cognitive and behavioral processes are based.42-51 Therefore, alterations to glia underlie pathological processes, and thus, it is logical that therapeutic interventions aimed at maintaining or improving their activity will help prevent or treat neurodegeneration.
There is already much support for the claim that studying glial alterations in AD will provide a basis to understand and treat this disease.23,48,49,52-57 However, unfortunately, we remain far from understanding the precise role of the different glial cells in each phase of every neuropathological process.3,26,58,59 Furthermore, many of the observations made in the human brain, as well as results obtained in animal models of AD, have generated some controversy about the true role (neuroprotective or neurotoxic) of the changes experienced by the different types of glial cells. Nevertheless, we do have enough information to establish that understanding the alterations to glia will shed some light on the mechanisms underlying different neuropathological processes; this information will certainly help to reveal therapeutic targets to fight these diseases. In this first part of this review, we consider the general characteristics of neuroglial cells in AD to understand their potential role in the pathogenesis, diagnosis and treatment (preventive or palliative) of this condition. We consider these issues in the light of differentiated and/or concurrent pathogenic and neuroprotective/neuroreparative processes. In the second part of this monograph (“The relationships between neuroglial alterations and neuronal changes in Alzheimer’s Disease, and the existing controversies. II Gliotherapy and multimodal AD therapy”) we will study the therapeutic possibilities that are being analyzed to try to prevent or palliate the onset or progression of this disease, based on treatments aimed at maintaining the normal functions of neuroglial cells or normalizing the alterations that occur in the progression of AD.
Some considerations about neuroglial cells
From Santiago Ramón y Cajal’s pioneering studies and those of his disciples and peers,19-22,27,41,60-69 we know that different types of glial cells accompany neurons in the central nervous system (CNS). These glial cells can be grouped into three main classes, initially characterized by their shape and their relationship with neurons: astroglia,3,19-26,61-66 oligodendroglia68,70,71 and microglia.27,28,58,67,69 However, as years have passed, we can now further differentiate these cells according to their functions, especially in terms of maintaining or not the activity of neurons and neural circuits or the production of different types of neuroactive substances (neurotoxic or neuroprotective astrocytes43,59 or microglial cells44-46 (see later).
Astroglia display a high degree of plasticity (glioplasticity), adapting to different situations in conjunction with their accompanying neurons to achieve the optimal degree of neuron functionality and the best response to normal and/or abnormal changes in the CNS.3,26 A significant advance in the study of astroglia has been understanding that some cell subtypes are able to produce active substances that protect neurons and contribute to neurotransmission (neurotrophic factors, neuroprotective substances, gliotransmitters, etc.). However, it was also found that reactive astrocytes can produce neurotoxic substances that induce neurodegeneration (cytokines, chemokines, free radicals, nitric oxide -NO-, prostaglandins, etc).23-26,35,42,43,47,53 In recent years, the study of astroglia has intensified with the application of new biochemical and molecular biology techniques50,52,53,73-87 and has revealed new characteristics and functions. As a result, in addition to the morphologically defined subtypes, new subtypes have been characterized through the expression of different genes in resting situations and as part of adaptive, neuroreparative, neuroprotective or neurotoxic responses.78-86 Single-cell transcriptomic and epigenomic analysis is rapidly becoming the method of choice to identify candidate regulators of cell identity and to distinguish the heterogeneity in neuroglial subsets. Such approaches enable developmental processes and disease responses to be studied more accurately.79-85
The most widely used marker of astroglia is GFAP. Its use has shown that there are many cells that clearly express this marker, and there are a number of cells that express it weakly and many that do not. Also, there are cells that contain different GFAP isoforms in both normal and pathological situations that are associated with hypertrophy and/or hyperplasia.86-88 Indeed, similar behavior has been observed with vimentin and other astroglia markers.80,81 Accordingly, CD44 is an extracellular marker of astrocytes, and CD44- and CD44+ cells have been identified in different subtypes of protoplasmic astrocytes, both with short or long extensions, and have been located in different layers of the cerebral cortex, hippocampus, and internal regions of the brain. 83 Likewise, other astroglia markers related to the metabolism of the neurotransmitter glutamate, such as glutamine synthetase or glutamate transporter-1,74,76,89 are present in different subtypes of normal and reactive astrocytes. These markers can be found in different regions of the CNS, both in relation to normal neuronal circuits and in areas of neurodegeneration in human brains, as well as in AD models. Perhaps the most important revelation in recent years has been the demonstration of the heterogeneity of astrocytes in relation to their location in the different cortical and hippocampal layers. Superficial, mid-, and deep astrocyte identities were recently identified in a pattern that reflected a gradient across layers that was distinct from that of neurons. 79 Features of these astrocyte layers were established in the early postnatal cortex, and these layers persist in the cortex of adult mice and humans. The presence of astrocyte layers in the adult cortex was confirmed through single-cell RNA sequencing and spatial reconstruction analysis, again establishing that these layers do not correspond to known neuronal layers. Both in rodents and in humans, these glial cells express genes whose functions have yet to be clarified, both in normal situations and during degeneration.
From the outset of studies on astroglia, different hypertrophic changes in some elements (together with cellular hyperplasia in some instances) were evident in response to aggressive agents (lesions, toxins, neurodegenerative processes or during aging19-21,41,59), as well as involutive processes (klasmodendrosis or klasmatodendrosis) in other cells19-21,41 (see later).
More recent studies of astroglia in these situations have shown how reactive astrocytes undergo phenotypic changes in two opposite directions: neurotoxic (A1) or neuroprotective (A2).3,26,47,48,52,59,74,77,78,90,91 In a study on gene transcriptome analyses of reactive astrocytes 92 was found that 57 genes (among others, C3, GBP2 -guanine nucleotide binding protein 2-, Serping1), were preferentially expressed in LPS-induced A1 reactive astrocytes, and 150 genes (S100a10 - calcium-binding protein A10- , PTX3 - pentraxin-3 -, S1Pr3, and others), were preferentially expressed in induced A2 astrocytes. C3 is, in many studies, the most commonly used specific marker for A1 astrocytes, while S100a10 and PTX3 are the most commonly used specific markers for A2 astrocytes.93-96
A1 reactive astrocytes seem to be mainly induced by reactive microglia (through TNF-α, L-1 and complement C1 subtypes).77,78 Different direct and indirect studies support this opinion: a) in triple knockout mice (IL-lα −/−, TNFα −/− and C1qa −/−), it has been observed that A1 reactivity decreased after systemic LPS injection, a model for neuroinflammatory research; b) this subtype of reactive astroglia is not generated in mice devoid of microglia (CSFR knockout mice).77,78,91,97 Another inflammatory cytokine, IL-18, effector cytokine processed by NLRP3 inflammasomes, 98 is a novel described inducer of A1 astrocytes, as well NO and oxidative products. These A1 inductors could downregulate the expression of A2 astrocytes. 99
A1 astrocytes can be identified in vivo by C3d complement upregulation. 58 Other suggested markers are H2-T23, Fkbp5 and ligp1. 99 These neurotoxic astrocytes can enhance local neurodegeneration or, conversely, they may help remove unrecoverable neurons in an attempt to repair neuronal circuits.78,90,91,97 A1 astrocytes also produce different neurotoxins not well identified until now, D-serine, NO and proinflammatory cytokines (TN-α and so on). 99 These substances, produced in different quantities in the different subtypes of A1 astrocytes, induce neuronal apoptosis and different alterations in oligodendrocytes (including pro-oligodendrocytes) and microglial cells. 99 Different local physiological or pathophysiological scenarios can reactivate the neuroinflammatory process that has started or that is ongoing, or, on the contrary, to initiate an anti-inflammatory response tending to restore normality of the CNS (although, unfortunately, it is the rarest possibility). TGF-β and FGFs have been reported to reduce neuroinflammation caused by activated microglia and astrocytes.100-102 Liddelow et al. (2017) further found that TGF-β1 downregulated several genes related to the A1 phenotype. A1 astrocytes can enhance local neurodegeneration or, conversely, they may help remove unrecoverable neurons in an attempt to repair neuronal circuits.78,90,91,97,103
A2 astrocytes are mainly induced by cytokines IL-1β 104 and IL10 (markers of anti-inflammatory M2 phenotype microglia). It is also notable the communication between M2 microglia and A1 astrocytes in antiinflammatory processes 105 because a reduction of A1 toxic molecules was observed. It was recently reported that TGF-β1 and BMP4 of the TGF-β superfamily signaling system modulated the plasticity of reactive astrocytes towards the A2 phenotype. 106 However, the main inducer of A2 astrocytes seems to be a chemokine. Prokineticin 2 (PK2), being neurons their major source.99,107 PK2 treatment or overexpression in primary astrocytes or the mouse brain can induce the reactivity of A2 astrocytes. 108 A2 astrocytes increase the production of neurotrophic factors, anti-inflammatory cytokines IL6 and IL10, and thrombospondins. A2 astrocytes are considered key elements in neuroprotection and the induction of synaptogenesis and neurorepair, as well as the main defense against neuroinflammation originated in microglial cells 99
Many results of studies on the induction of A1 and A2 astrocytes, as well as the production of neurotoxic or neuroprotective substances derived from these astrocyte subtypes, give rise to controversies that are difficult to solve. Therefore, it is necessary to intensify research in various fields and models (molecular, cell and animal models) to reach valid conclusions.99,109 In a recent review on the different types/subtypes of reactive astrocytes, a consensus (subscribed by 78 researchers) is proposed to define subtypes based on different markers (morphology, location, genetics, RNA expression, neuronal and glial effects) in order to clarify the subtypes of astrocytes and their specific role in glioprotection and gliopathology. 59
Different subtypes of non-motile myelinating oligodendrocytes (OL) with a distinct stellate morphology, with fine processes in parallel or connected to myelinated internodes, have been described. Other classifications have been based on the number of their enveloping processes of neuronal axons and the characteristics of these axons.68,71 Oligodendrocytes undergo many maturation states before reaching their destination and becoming a mature myelinating OL. 116
OPCs in the brain are produced in three distinct waves (from embryonic to postnatal ages) and from distinct regions (the ventral ventricular germinal zone and the dorsal aspect of the spinal cord), but only OPCs and their progeny from the last two waves persist until adulthood. OPCs from different regional origins have diverse properties (i.e., the dorsally derived OPCs have a higher remyelination capacity. 117 The oligodendrocyte-specific Gprotein-coupled receptor GPR17 is a cell-intrinsic timer of myelination. 118 The study of OPC properties have revealed that these cells are indeed not a uniform population with equal behaviors or functions. OPCs in different regions show different responsiveness to growth factors or mitogens (OPCs in the white matter, but not in the grey matter, respond to the platelet derived growth factor -PDGF - 119 ) and vary in their capacity to differentiate when transplanted into other CNS areas.120-122 However, it remains unclear whether the reported diversity of OPC properties represent subtypes of OPCs with distinct functions, or if they reflect different states of cells with the same function as they progress along their lineage. 116 Very different phenotypes have been described in different studies, but no clear subtypes related with CNS regions, age or pathological states.120,121 This cell subtype appears to proliferate and enter damaged areas of the CNS, accumulating to the greatest extent in areas with neurodegenerative lesions.123-126 In a recent study, senescent pro-oligodendrocytes were found to associate in large numbers with plaques in both AD models and human brains.110,123,126
There are various morphofunctional subtypes of microglia, with a round morphology (predominantly phagocytic) or with extensions,27,28,34,44,46 contributing to the main line of immune defense in the nervous system.137-140 Microglia exhibit regional and age-dependent phenotypes,141-143 and their coupled proliferation and apoptosis maintain the rapid turnover of microglia in the adult brain, 128 mainly in changes of the normal nervous tissue status. Reu et al, 2017, report that microglia, unlike most other hematopoietic lineages, renew slowly at a median rate of 28% per year, and some microglia last for more than two decades. Microglia population in the human brain is sustained by continuous slow turnover throughout adult life.128,144
Microglia are highly reactive cells (many types of “activated”, “reactive” or “hypertrophic” microglial cells have been described). Activated microglia present an increased number of amoeboid forms and decreased arborization in cells with prolongations, 145 and typically are distinguished by overexpression of human leukocyte antigen-antigen D related (HLADR) as well as a group of cluster of differentiation (CD) molecules such as CD40, CD45, and CD68.146-148 For many years, reactive microglial cells were considered to exhibit two opposite states of proinflammatory/neurotoxic substance production (proinflammatory cytokines, such as IL-1α, IL-1, IL-6, IL-12, IL-15, IL-17, monocyte chemoattractant protein-1 -MCP-1-, and TNF-α; reactive oxygen -ROS - and nitrogen -NOS- species) or anti-inflammatory/neuroprotective (anti-inflammatory cytokines, including IL-10, IL-4, transforming growth factor-beta -TGF-growth factors).145,149 Thus, two phenotypes were, respectively, distinguished: M1 and M2 were termed. 19 All these substances are close related to the induction of diverse reactive astroglial cells (see astroglia section). These two main classes of reactive microglia (M1 and M2) do not develop in clearly specific brain regions or disease phases.141-143,150-152 Indeed, markers for both populations can be either up- or downregulated in many different areas in the brain in association with a variety of neurodegenerative diseases.150-153 New technologies (including two-photon imaging, whole-genome transcriptomic and epigenomic analysis with complementary bioinformatics, unbiased proteomics, cytometry by time of flight (CyTOF; Fluidigm) cytometry) need to be used to define different subtypes of microglia reactive cells. 151 A comprehensive gene expression encyclopedia of glia cells exists to help researchers 154
Ransohoff (2016) 151 raises an important question (A polarizing question: do M1 and M2 microglia exist?), considering that this microglial polarization has not been established by research findings, being adopted this nomenclature in an attempt to simplify data interpretation at a time when the ontogeny and functional significance of microglia had not yet been characterized.
Microglial cells can communicate with other cell types by releasing soluble factors as well as exchanging active molecules an RNA through secreted extracellular vesicles (EV)/exosomes, ranging in size from 30 to 100nm, and diffusing over long distances. 155-157
Microglial cell reactivity is critical in neuroinflammation and is closely related to most of the neuropathological cascades that lead to dementia.138-140,158-160 However, microglial dystrophy has been correlated with the presence of neurofibrillary degeneration in situ, suggesting that neurodegeneration is secondary to age-related microglial deterioration. 161 Microglial dystrophy has been detected in the human brain,129,162 and the term “dystrophy” is used to refer to different morphological abnormalities that affect cytoplasmic microglial extensions, such as spheroid swellings, deramifications, beaded or tortuous processes, and—most conspicuously—fragmented processes. This latter phenomenon probably represents the most advanced stage of cytoplasmic deterioration that affects microglia, and the term “cytorrhexis” has been proposed to describe specific microglial cytoplasmic fragmentation. 129
Neuroglial Responses in AD
Astrogliosis and microgliosis are key aspects in most theories regarding the pathogenic “cascades” leading to AD,3,24-28,31,33-35,138-143,150-153,158-161 although other changes in astroglial, oligodendroglial and microglial cells have also been described81,91,110,125,126,159,163-165 but have not been thoughtfully considered in the neuropathological theories on AD.
Most neuroglial studies have been carried out using only a limited number of specific markers for certain types or subtypes of neuroglial cells (normal and/or reactive). The results of these studies have been considered useful for a sufficient definition of the neuroglial typology of the region/area studied in the brain. However, many astroglial cells are not GFAP- or vimentin-immunopositive, and many microglial cells are not IBA-1 or LN-3-immunopositive. As an example, Figure 1 shows a high density of neuroglial nuclei in an area of cerebral cortex from a case of AD using a general stain (Congo red in this case) to visualize cells. If the density of different types and subtypes of neuroglial cells (astrocytes, oligodendroglial cells, including NG2+ cells, and microglia) is studied in parallel sections with the most commonly used conventional markers in research, the sum of the partial results obtained from each neuroglial type/subtype is always lower (17-32% in our studies, pending of publication) than the density of neuroglial cell nuclei. An important number of neuroglial cells of specific phenotypes will go unnoticed. Brodmannn's area 46 of a brain from an AD case, Braak and Braak V. Layer IV-V. Zone of high density of glial cells scattered in the cortical parenchyma and not related to amyloid plaques. Congo red stain / confocal observation with green filter. A large number of neuroglial nuclei (from astrocytes, oligodendrocytes, and microglial cells) is observed. The technique does not allow to differentiate the different types or subtypes of neuroglial cells, but it demonstrates the high density of neuroglial cells that are present in this region of the CNS altered by AD pathology. Bar = 200 microns
It is true that there are different brain areas where important conventional astrogliosis and/or microgliosis processes can be observed (see later). However, there are also areas where there is a reduction in astrogliosis or microgliosis (Figure 2) or an abnormal manifestation of astroglial cells presenting unusual markers (such as amyloid in astrocytes; (Figure 3) that are not commonly considered. New techniques are indispensable for studying well-characterized glial cells in different regions/areas of the brain.79-85 Brodmannn's area 46 of a brain from an AD case, Braak and Braak V. Layer II / III. Area of low incidence of glial cells. Bielschowsky silver impregnation. The intensely stained nuclei correspond mostly to microglial cells and are largely associated with amyloid plaques, both with and without “core”. Bar = 150 microns Brodmannn's area 46 of a brain from an AD case, Braak and Braak V. Layer I / II. Area of low density of neuroglial cells, especially astrocytes. Section immunostained with amyloid antibody 6E10 plus hematoxylin contrast. Amyloid plaques and small deposits of intraparenchymal amyloid are observed. Many of the astroglial cells (arrows) (confirmed in parallel sections immunostained with GFAP antibody) show amyloid reaction. Bar = 150 microns

The main relevance of neuroinflammation in AD pathogenesis is indisputably accepted. In this sense, the differential involvement of neuroinflammatory molecules, mainly released by microglial cells during the development of the disease, may contribute to the modulation of characteristics and the severity of the neuropathological changes, driving, in part, AD phenotypic diversity. 58 Amyloid production and deposition are closely related to neuroinflammatory reactions of astroglial and microglial cells. Moreover, astrocytes and microglial cells (in different transitional states) seem to have an important involvement in tau production, degradation, processing and propagation.166-168 However, we still do not have a clear idea of the different toxic or neuroprotective mechanisms that actually occur. The ability to interpret the effects of all these neuroglial changes is hindered by the fact that the exact role of these glial cells in AD is not fully understood. Indeed, most neuroglial alterations, such as gliosis, are also considered processes inherent to normal CNS aging, and they have been associated with other neurodegenerative diseases. Therefore, many mysteries remain regarding the influence of glial cells on the pathogenesis of AD.3,23-26,153,158 Indeed, it is still unclear whether glial modifications or their reactive states fulfil a primary or secondary role in the “cascades” or events that drive neurodegeneration.
Importantly, there is no single relationship between the different types of glial cells and neurons affected by accumulations of aberrant proteins (phospho-tau, synuclein) (Figures 4A, 4B) or the amyloid plaques (Figures 5, 6 and 7). In fact, different types of amyloid plaques are not associated with specific patterns of glial accompaniment. The number of astroglial cells closed involved varies between 2 and 15 (Toledano and colleagues, unpublished data) (Figures 5, 6 and 7), with neighboring similar plaques also displaying considerable variability (this was observed in all regions of the brain, regardless of the number of astrocytes in each studied area). Many plaques are devoid of astroglial accompaniment, in special in areas of low astroglial density (astroglial involution?). Microglia seem to be more consistent in their relationship with amyloid plaques, as microglia are evident with more than 75% of all plaque types, both in their mass and in the periphery. A and B. Brodmann’s area 46 of a brain from an AD case, Braak and Braak IV. GFAP immunostaining plus hematoxylin staining contrast. Fig. 4A. Layer V. Zone of high density of hypertrophic GFAP hyper-reactive astroglial cells scattered in the cortical parenchyma, mostly related to dystrophic neurons but not to amyloid plaques. A large number of glial nuclei (oligodendrocytes and microglial cells, as well as GFAP immunonegative astroglial cells) is observed. Fig 4B. Small area of this zone where hypertrophic astrocytes are observed in the process of klasmatodendrosis, with a great fragmentation of their glial extensions. Bar, Fig 4A = 50 microns; Fig 4B = 40 microns. Astroglial cells (GFAP immunostaining) in a case of AD, Braak and Braak IV. CA1 region of the hippocampus. Highly complex amyloid plaques with a variable "crown" of hypertrophic and hyperreactive astrocyte cells. Astrocytic extensions barely penetrate the plaques. (Hematoxylin contrast). Bar = 100 microns. Brodmann's area 46, layer I / III. Different types of clusters of astroglial cells randomly dispersed and with little relation to amyloid plaques (revealed in a parallel section) (without H / E contrast). Bar = 200 microns . Brodmann's area 46, layer V. High incidence of hypertrophic and hyperreactive astrocytes surrounding a large amyloid plaque and relating to vessels and neurons of normal appearance in this layer. Bar = 100microns.



Glial alterations must always be considered when attempting to understand the pathogenesis of neurodegenerative diseases, especially AD, and when attempting to develop successful therapeutic strategies. In fact, glial cells are currently considered to represent a promising target to establish effective therapies for AD, mainly because other therapies targeting neurons have failed to produce promising results.
The special features of neuroglial cells in human AD
Astroglial responses
There is some debate as to whether astrocytes display any special features in AD. Unusual expression of GFAP isoforms has been described,87,89 and specific patterns of astroglial reactivity have been proposed to be closely associated with certain pathological AD lesions or specific areas of amyloidogenesis, but this is a debatable matter (Figures 1, 2, 3, 4, 5, 6, 7, 8 9, 10, and 11). Small foci of hypertrophic astrocyte clusters - hyperreactive GFAPs, unrelated to amyloid deposits and / or dystrophic neurons, in one case of AD, Braak and Braak, III, Brodmann's area 46. GFAP immunostaining plus hematoxylin contrast. No amyloid plaques were demonstrated in parallel sections. Bar = 40 microns. Small foci of hypertrophic astrocyte clusters - hyperreactive GFAPs, unrelated to amyloid deposits and / or dystrophic neurons, in one case of AD, Braak and Braak, III, Brodmann's area 7. GFAP immunostaining plus hematoxylin contrast. No amyloid plaques were demonstrated in parallel sections. Bar = 40 microns. Cerebellum (vermis, lobe VI) of a case of AD, Braak and Braak, III. GFAP immunostaining. Hyperreactive normal and hypertrophic / GFAP astroglial cells are observed, both stellate and elements of the Golgi epithelial glia, as well as numerous astroglial nuclei of hyperplasic GFAP immunonegative astroglia. Bar = 150 microns. Cerebellum (vermis, lobe VI) of a case of AD, Braak and Braak, III. Nitrotyrosine immunostaining (degenerative reaction marker). Staining is observed in most of the hyperplasic cells in the Purkinje cell layer. Bar = 150 microns.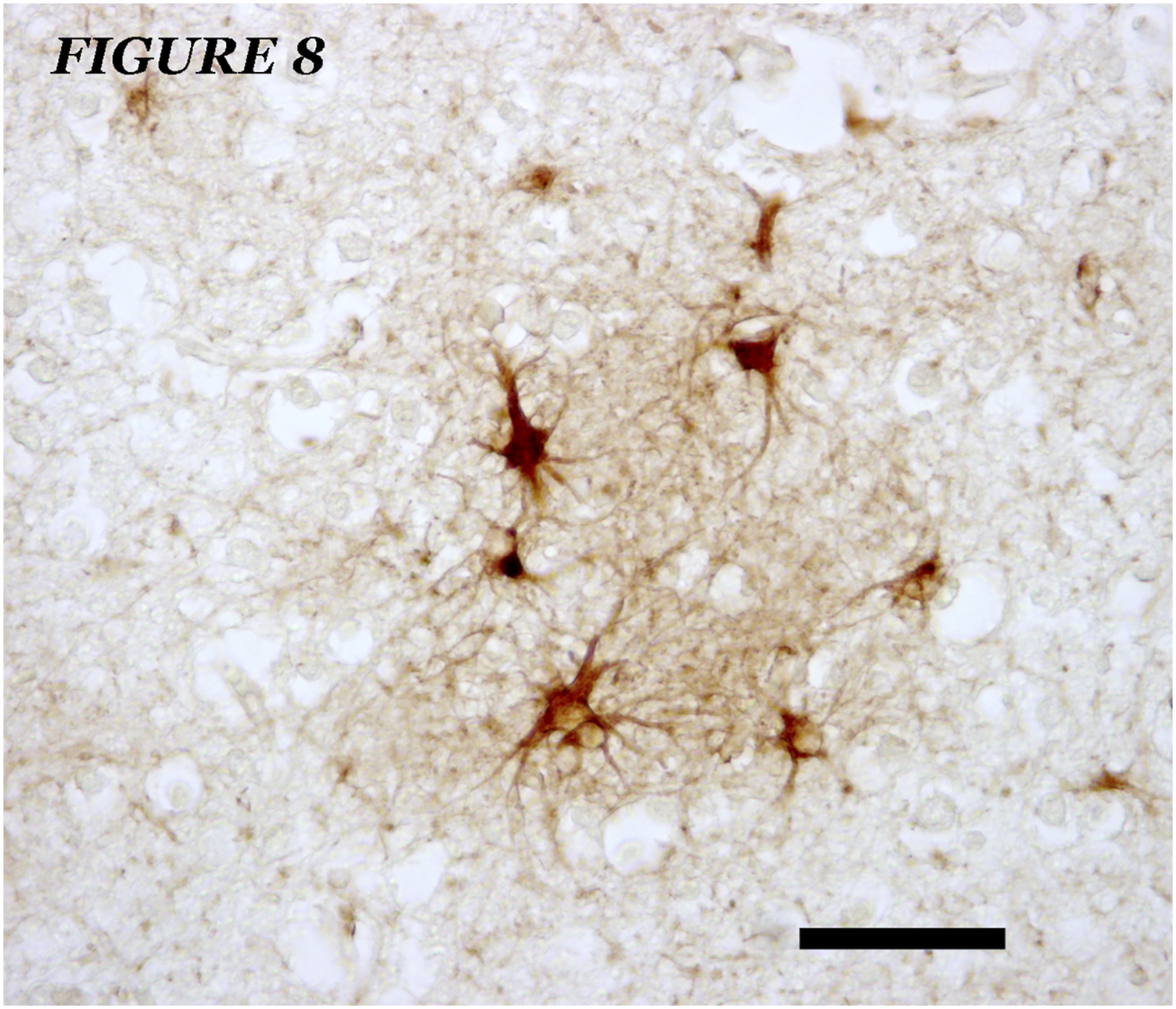



“Dramatic” and “generalized” astrogliosis has been described in AD,87,169 and a wide number of hypertrophic/GFAP-hyperimmunoreactive astrocytes have been described in many regions of the brains of AD subjects. Moreover, special patterns of astrogliosis have been observed in some specific CNS regions, but do not always occur. It has been proposed that cortical astrogliosis in AD follows an obvious laminar pattern, with a heavy band evident in layers I-III and another band present in layer V 160 (Figures 2, 3, and 4), even though this pattern has not been reported universally. Reactive astrocytes have been reported close to amyloid (simple or complex) plaques in the hippocampus (Figure 2). Normal aging is sometimes associated with considerable cortical gliosis but does not follow any specific pattern.26,170,171 Studies of astroglial markers show a complex age-dependent remodeling of these cells in different brain regions, 86 although in most AD cases, the hypertrophy of immunoreactive astrocytes in the gray and white matter is heterogeneous (in some cases, hypertrophy is more pronounced at the interface between the two types of matter). Subcortical gray matter astrogliosis is commonly observed in both normal aging and AD, although “variable” and “disorganized astrogliosis” is more often observed in AD, 86 even in areas where astroglial cells are lost to a large extent 86 (Figure 6). Such areas have been considered zones in which senescence, atrophy or dystrophy of these cells occurs, provoking neuronal pathologies; the maintenance of normal astrocytes is necessary for the normal activity of neurons and their adaptive changes.24,52,162
Several studies suggest that the number of astroglial cells in many regions of the CNS remains (more or less) constant throughout life, irrespective of the evolution of pathological processes such as AD.40,161,165,172 Phenotypic changes but not proliferation have been proposed as glial responses to AD. 172 We recently found that astrogliosis (defined by the distribution of GFAP) in the brains of 65- and 85-year-old AD patients (with a disease course of 10-15 years) was less pronounced than that observed in normal 85-year-old individuals. 86 Astrogliosis in AD patients seems to be quite variable in different brain regions and reflects the state of involution in each brain. Astroglial GFAP-immunopositive cells and immunonegative hyperplasia (documented by the increase in astroglial nuclei and the absence of microglia) are the sum of both general and local reactions to neural changes; these changes reflect the status of the environment. In neurodegenerative processes, neuronal and astrocyte death, as well as some hypertrophic surviving astrocytes, seem to compensate for the loss of astroglial subtypes, as demonstrated in the cerebellum or in the molecular layer of the hippocampus. 86
Despite these considerations, astrogliosis is thought to be associated with neuropathological alterations in AD, and changes in the number of astrocytes are associated with amyloid plaques in several studies. The significance of reactive astrogliosis around these pathological structures is unclear (noxious or protective)3,25,59,161 and different plaque-astroglial cell relationships have been observed: plaques with a scar-like crown of astroglia, plaques with isolated peripheral astrocytes, plaques infiltrated by astroglial processes, plaques not associated with astroglia, etc. A generally weaker astrocyte response to β-amyloid (Ab) plaques has been associated with cognitive impairment (perhaps a loss of defense responses),173-175 and reactive astrogliosis in the neuropil of affectated areas is considered to be harmful. In a recent study, the highest neurotoxic plaques were related to reduced contact with astrocytes.175,176 In terms of neurofibrillary tangles, an association between neurons with tangles and glial cells was observed.20,26,176 Indeed, there appears to be a clear relationship between such neurons and reactive astrocytes,19,20,26,161,176 although different types of glial-neuron relationships have been described.
We present a series of images showing different reactive astrocytes (GFAP immunopositive) related to neuropathological alterations (or possible foci of alterations) in AD subjects which are not generally considered in current studies. In Figure 4A, a zone of high density of immunopositive GFAP astrocytes is shown, mainly associated with dystrophic neurons and not associated with amyloid plaques. In some areas of this brain region, diffuse involutive hypertrophic astrocytes are evident that undergo a process of klasmatodendrosis (degeneration of the cell body and its processes) (Figure 4B). This process could be a transition state towards astrocyte involution. In some other areas where there is no marked AD neuropathology, foci of astroglial hypertrophy (Figures 8 and 9) similar to those that appear in advanced AD brains (included areas of astrglial involution – Figure 6) can be seen and may be indicative of a zone of degenerative onset.
Astrocytes that accumulate amyloid or aberrant tau protein deposits have often been found in human AD patients (Figure 3), in experimental AD models and in animals that present AD-like amyloidosis (monkeys and simians).177-180 Moreover, astroglial cells may generate amyloids, raising the possibility that astrocytes participate in the generation of amyloid that leads to AD. Extracellular vesicles from 3xTgAD mouse and AD patient astrocytes have been shown to be transporting elements of substances that cause damage to neurons, glial cells and endothelial cells, aggravating AD 181 Hypertrophic astrocytes with abnormal gene expression, such as calretinin, have also been described. 86 Indeed, there is an increase in the GFAP 1 isoform, one of the nine splice variants of GFAP described in astrocytes from different species, as AD progresses.87,88 Complex and region-specific changes to other astroglial markers (glutamine synthetase, S100β) have also been detected in both the aging brain and in AD.89,163 Thus, the diverse astroglial responses in the brains of AD patients could reflect the multifactorial nature of this disease (a systemic disease that alters glial responses in different ways3,19,170,182), although these responses may also reflect the particular features of the distinct CNS regions.24,26,86
Oligodendroglial responses
Alterations to oligodendrocytes have rarely been studied in AD, although demyelination is a secondary characteristic feature of this disease. Demyelination assessment using new brain imaging techniques is widely used for in vivo diagnosis,183-186 although demyelination is mainly considered a secondary pathological event in AD. As such, more studies are needed to assess whether demyelination is a primary pathological process in AD. Metabolic changes in AD oligodendrocytes have been observed, such as alterations in glycolytic and ketolytic gene expression. 187
In recent years, much interest has been generated by NG2+ cells.71,112,126,188 These cells seem to be modified in AD subjects, even though the significance of these alterations (neuroprotective or neurodegenerative) is not completely clear.
Microglial responses
There is an intimate relationship between microglia and neurons at the synaptic level. Microglia modulate activity-dependent functional and structural plasticity indispensable to normal synaptic function and cognition. Alterations in microglia-synapse interactions are key for AD presentation and progression. 189
Different reactive microglia have been described in association with neuropathological AD lesions, whereas less prominent differences in these cells are associated with normal senility or with other neurodegenerative diseases. Microglial cells with abundant extensions, scattered though the parenchyma, have been observed in different regions of brains affected of AD (Figures 12 and 13). Different morphologies have also been described in other areas of these brains. The forms with extensions also invade some subtypes of amyloid plaques and the round forms are close the plaques as well diffusely distributed in the brain parenchyma (Figures 12, 13, and 14). Different microglial phenotypes have been described,127,130,138,140,141,158,160 but no clear microglial responses have been associated to specific alterations in specific areas of the brain or in specific phases of the progression of AD Microglia cells in a case of AD, Braak and Braak III, Brodmann's area 46, layer IV / V. IBA-1 immunostaining. High density of microglial cells with abundant extensions that are scattered throughout the parenchyma. Bar = 50 microns. Microglia cells in a case of AD, Braak and Braak III, Brodmann's area 46, layer IV / V. Lectin immunostaining. High density of microglial cells, both with rounded morphology and with extensions, scattered throughout the parenchyma. The forms with extensions invade the amyloid plates. Bar = 50 microns. A and B. Microglia cells in a case of AD, Braak and Braak III, Brodmann's area 46, layer IV / V. Silver impregnation, Bielchowsky block method. Amyloid plaques ((Bar = A, 65 microns and B, 50 microns of diameter) without core (A) and with core (B) showing microglial invasion.


A decrease in ramified (considered healthy) microglia was recently described in subjects with advanced AD, 130 with imaging analysis demonstrating a reduction in the arborized area and skeletal complexity. It was concluded that activated microglia were not associated with AD but that they were increased in nondemented controls with a stronger AD-type pathology. Moreover, the authors considered that microglial clusters were only occasionally associated with Aβ- and tau-positive plaques but that these elements represented less than 2% of the total microglial population. We have shown that the number of microglial cells in the cerebellum and hippocampus is more closely related to the age of the individuals than to their AD pathology. 86
Neuroinflammation is currently thought to be one of the main mechanisms driving the pathogenesis of AD,3,26-28,33-36,40,164,168 although this process can also explain degenerative changes in aging and in other neurodegenerative processes.3,26,27,34,168 Microgliosis could protect against neuronal degenerative changes (via phagocytosis of damaged neurons or amyloid plaques 190 or via the production of neuroprotective agents 191 although a secondary effect might provoke alterations through an excess of neurotoxins in the neural microenvironment (e.g., chemokines and cytokines or inducers of oxidative stress).140,164,192-196 A large increase in both resident and reactive microglial cells is often observed in the parenchyma, as well as around the vessels in AD brains (Fig 12). Microglial activation in AD appears to be Aβ-dependent, with Aβ binding to receptors such as RAGE, scavenger receptors 144 and toll-like receptors (TLR2, TLR4 and TLR6) 197 representing an important cause of microglial activation in mouse models of AD. However, clear patterns of microgliosis are not generally evident in AD, although a higher density of microglial cells in subpial zones, as well as at the transition between the neuronal and molecular layers of the hippocampus and cerebellum, has often been seen. 66 Many authors consider that microglial cells accumulate in areas more strongly associated with amyloid deposits, even infiltrating amyloid plaques, although they also appear in less severely affected areas.198,199 In some models of AD, microgliosis is observed before the onset of AD pathology, which is why inflammation/microgliosis has been considered the origin of AD in some theories. Significantly, Aβ immunotherapy seems to downregulate microglial activation and reduce the inflammation-mediated component of AD. 190
Despite microglial reactions, the genotype of microglial cells in different individuals is closely related to AD progression. Microglial TREM2 (Triggerin Receptor Expressed on Myeloid cells 2) facilitates adaptive regulation of amyloid plaque formation; as amyloid fibrils become compacted into plaques, adaptive regulation reduces the local induction of neurodegeneration due to the TREM2 activity of healthy microglial cells. However, other TREM2 variants, such as the R47H variant, are associated not only with a higher risk of AD but also with earlier symptom onset and accelerated dementia.200-206 Other single-nucleotide polymorphisms in genes that are exclusively or largely expressed in microglia, including CD33, CR1, ABCA7 and SHIP1, are associated with an increased AD risk. 152 Human and mouse single-nucleus transcriptomics revealed TREMP2-dependent and TREMP2-independent cellular responses in AD, 189 and TREMP2 haplodeficiency impairs microglial barrier function, decreasing amyloid compaction. 207 In this sense, colony-stimulating factor 1 receptor signaling seems to be necessary for microglial viability. 207
Microglial dystrophy has been demonstrated to be correlated with the presence of neurofibrillary degeneration, suggesting that neurodegeneration is secondary to aging-related microglial deterioration.161,162 Dystrophic microglia are also associated with different neurodegenerative diseases. 208 Moreover, “aged microglia” seem to affect synaptic function. 209 The inability of microglia to remove amyloid deposits has been considered a cause of “microglial exhaustion”, which in turn promotes neurofibrillar neurodegeneration, brain failure and dementia. 161
It has been suggested that the inability of microglia to remove aggregated amyloid causes microglial exhaustion and thus exacerbates already ongoing age-dependent microglial deterioration. 129 The eventual total loss of functional microglia in advanced AD could promote widespread NFTs, dementia and brain failure. Microglial dystrophy is probably caused by oxidative stress, and in this sense, it can be considered pivotal in this disease.138,161,162
In summary, there is structural evidence of microglial heterogeneity in human AD from light microscopy and transmission and scanning electron microscopy in AD models in association with amyloid and tau pathology,130,198 as well as molecular evidence of the production of a large variety of neurotoxic substances. The sequence of appearance of the microglial reaction and that of the disappearance of normal microglia in human AD has been discussed in various studies certain studies show microglial alterations in nonsymptomatic or early AD phases and others only show microglial alterations in advanced phases. The loss of healthy microglia has also been described only in severely affected regions of AD brains. 130
Chronically activated microglia secrete proinflammatory cytokines related to the induction and/or progression of AD, but the state transition from a resting state to an activated state and the exact meaning of each phase (depending on the panel of cytokines/chemokines secreted) remain unclear. 210 Natural beneficial effects can occur, or therapeutically induced effects can be produced in this state or during this period of transition, but careful research is necessary.
Induced glial responses in AD models
In some experimental models of AD, such as those involving mechanical, anoxic or toxic damage to the cortical regions involved in cognitive functions (e.g., the entorhinal cortex), 199 significant changes in resident neuroglial cells are induced (astroglia and microglia), both in local lesion areas and in areas innervated by injured neurons (e.g., the hippocampus).211,212 Similarly, damage to cholinergic cells in the nucleus basalis magnocellularis (nbm) 211 of 4-month-old rats causes transient changes in “proximal” areas, e.g., nondamaged structures neighboring the nbm that are not innervated by this nucleus but that maintain a vascular relationship with it; this damage also causes substantial and permanent changes in the ipsilateral cortex to which it is directly connected synaptically (layers I-V of the motor and somatosensory cortical regions). Moreover, the indirectly connected contralateral cortex displays long-term reactive astrogliosis, a cortical alteration that persists for relatively long periods (13-20 months). In contrast, the proximal response lasts from 1 day to 13 months, and tends to disappear thereafter. Tightly interwoven subsets of astrocytes with distinct GFAP immunoreactivity have been observed, while nbm lesions in 20-month-old animals produce similar but weaker patterns of glial reactivity, in addition to glial reactivity related to old age. The maintenance of reactive astrocytes for many months after the occurrence of a lesion suggests an influence of factors other than those produced by nbm neurons that were initially damaged. It is possible that similar reactive astrocytes in humans could promote AD-related neurodegeneration and that nbm cholinergic involution might provoke cortical involution by inducing reactive astrocytosis and/or microgliosis.
Reactive processes of different sets of neuroglial cells have been described in different transgenic mouse models of AD. In some cases, such glial alterations have been described early in life and prior to the appearance of any neuropathological hallmarks of AD, while others develop after the appearance of aberrant deposits of amyloid or tau protein162,163 (Figure 15). Enrichment of the neurodegenerative signature in microglia has been observed in AD models.
213
Electron microscopy image of an amyloid plaque of the frontoparietal cortex (layer V) in a transgenic model of AD (APP + PS1, to which two human genes have been inserted - Amiloid Precursor Protein and Pre-Seniline 1- that induce AD of family type). In the center of the image, an amyloid plaque is observed, with a dense amyloid “core” and less electrodense radial amyloid extensions. The boundaries of a hypertrophic astroglial cell are marked in red, and the extensions of a microglial cell associated to the amyloid plaque are marked in blue. In black, hypertrophic neurites filled with vesicular forms indicative of degeneration of dendrites and axons of affected neurons are delimited. The almost “normal” appearance neuropil shows small alterations compared to control mice (increase in diameter and alterations of subcellular structures in dendrites and axons; synaptic alterations; varicosities in neuroglia extensions). Bar = 25 microns
How can neuroglial alterations be interpreted in human AD tissue and AD models?
Morphological and functional changes to neuroglia are always evident in the brains of humans suffering from AD, as well in the brains of animal models of AD, although these changes are quite variable in nature (Figure 15). These neuroglial variations can be interpreted in different manners. It may be that the main controversy, when one wants to interpret the role of astroglia and microglia associated with the neuropathological manifestations of AD, is whether the associated glial reactions tend to eliminate aberrant protein accumulations and/or to separate them from the rest of the tissue or, on the contrary, if these cellular elements promote further development of neuropathology. In all the studies on this subject, and in all the reviews on it, this controversy is always considered by all authors, and an undisputed conclusion is never reached. Neuroprotective and neurotoxic glial reactions seem to coexist. We still do not have markers (or a set of markers) that can be defined, with absolute certainty, regarding the role of each of the neuroglial cells present in certain regions of the CNS in each phase of a disease. Perhaps the most important justifications of the involvement and the simultaneous or exclusive neurotoxic and/or neurodegenerative effects of neuroglial cells in AD are indicated below. 1) Differences in the response of different neuroglial families or subtypes.
The plasticity of glial cells, particularly in terms of reactive gliosis, includes modifications to the structure of glial processes, changes in cell motility and—more importantly—modifications in the production of diverse neuroprotective (neurotrophic factors, gliotransmitters, etc.) and neurotoxic substances (cytokines, chemokines, free radicals, prostaglandins, NO or other neurotoxins).160,165,192 The expression of different GFAP isoforms in astrocytes, 87 as well as the different reactive forms of these normal neuroglial cells (or subsets of astroglia) 59 must also be kept in mind. Chronically activated microglia secrete proinflammatory cytokines related to the induction and/or progression of AD, but the state transition from a resting state to an activated state and the exact meaning of each phase (depending on the panel of cytokines/chemokines secreted) remain unclear. 210 These changes alter the concentrations of local factors, which could in turn induce neuropathological changes in small areas that may hinder the primary neuroprotective response driven by neuroglia. 86 Neurotoxic effects are commonly described, but natural beneficial effects could occur. Moreover, therapeutically induced effects could be produced in these state transitions. Careful research is necessary both to correctly interpret all neuroglial changes and to develop protective or corrective glial therapies.
2) Differences in pathological neuronal degenerative changes in AD.
An important number of studies on neurodegeneration in AD focus only on neuronal tau-dependent neurofibrillary tangles and dense core of amyloid plaques. However, other well-demonstrated pathological alterations, such as the existence of different types of dystrophic neurites, other aberrant protein aggregates in neurons and different types of amyloid deposits (diffuse or focused amyloids, which forms various types of plaques), are overlooked. All these alterations are related to neuroglial changes of varying intensity and make different contributions to the development of the disease. The alterations occur differently in each region/area of the brain parenchyma depending on the characteristics of the initial cellular or molecular changes.
AD is a multifactorial syndrome, both in its genesis and in the consequences of its different pathological modifications. In this line of thought, Fiala (2007) 214 pointed out several hypotheses that try to explain the local production of amyloids and the pathogenesis of the plaques: 1) a vascular (extracerebral) origin of amyloids leading to perivascular deposits and synaptic/dendritic amyloid lesions; 2) a glial origin, inducing neuropil deposition and synaptic/dendritic amyloid lesions; 3) neuronal secretion, glial activation and glial neurotoxicity; 4) neuronal secretion, glia-induced amyloid “fibrillation” in the neuropil, and synaptic/dendritic amyloid lesion; 5) Abeta release by neuronal lysis, deposit production and activation of microglia, and glial toxicity; and 6) dystrophic axon lysis (including amyloid spread), extracellular amyloidosis, glial reaction, neuronal and glial toxicity. Likewise, there are various hypotheses that assume that the hyperphosphorylated tau protein and other aberrant proteins give rise to neurite dystrophy and neuronal dysfunction or death. Astrocytes and microglial cells (in different transitional states) seem to have an important involvement in tau production, degradation, processing and propagation, but we still do not have a clear idea of the different toxic or neuroprotective mechanisms that actually occur 166,214
Expression and secretion of ApoE isoforms in astrocytes and microglial cells during neuroinflammation processes are of special importance in neurodegeneration. 215 ApoE, the main member of the apolipoprotein family in CNS, is a protein involved in a wide variety of functions, including lipid transport, neuromodulation, neuronal plasticity, neuronal repair, neurite outgrowth and regulation of Aβ formation and clearance.216,217 ApoE also modulates the inflammatory response of microglia and astrocytes, the cells that secrete the greatest amounts of this apolipoprotein.218,219 Three major isoforms, apoE2, apoE3, and apoE4, encoded by the ε2, ε3, and ε4 alleles, exits in humans. These isoforms have different abilities to carry out the assigned functions in the CNS. Apoe4 not only increases an individual’s risk for AD,220-222 but also for the outcome from neurological injuries with dramatic brain inflammation.221,223 Conversely, Apoe2 and 3 seem to be protective factors, 221 increasing anti-inflammation. 224 APOE4 genotype was associated with lower levels of secreted apoE from astrocytes and microglia, as well as higher levels of the larger apoE species that remained inside microglia. In neurons, apo 2 and 3, and apoE4 have different intracellular trafficking profiles: apoE4 is retained in the endoplasmic reticulum (ER) and Golgi apparatus, causing functional disturbances. 225 In astrocytes, apoE4 causes ER stress. 226 In studies carried out on cell cultures of astrocytic microglial cells that express different apoe2, 3 or 4 isoforms 215 it has been observed that APOE2 astrocytes and microglia secreted three to five times more apoE than APOE4 cells, with APOE3 cells intermediate, supporting the hypothesis that APOE4 predisposes to greater inflammation in primary cells. The presence of apoE4 is associated with overactive proinflammatory phenotypes such as elevated NO, TNFα, and IL-6.227,228 However, expression of astrocytic apoE3 decreased the levels of IL-6 and IL-1β. 229 The studies of Liu et al., 2017 229 conclude that expression of apoE4 during the initial seeding stage of AD is sufficient to drive amyloid pathology and plaque-associated neuritic dystrophy, while the presence of apoE4 after the initial seeding stage has minimal impact of amyloid pathology, highlighting the importance of early alterations of apoE in AD.
The above mentioned pathological alterations (tau and amyloid related alterations, apoe 4 dysfunctions) may be the origin of the varying neuronal changes and development of AD and should be considered when trying to interpret the role of neuroglia in specific regions/areas in the study of each brain.
This complex neurodegenerative process considered here involves the accumulation of products in the parenchyma, the astroglial and neuronal secretion of amyloids, amyloid fibrillation by microglia, neuronal lysis and of dystrophic axons, etc. Different compounds can be demonstrated in the plaques. Reactive subtypes of microglia and astroglia are involved in all these pathological processes, although their roles seem to differ in terms of the formation and development of the distinct types of plaque. Different subtypes of neuroglial cells are expected to be observed. Plaques do not grow indiscriminately because neuroglial cells regulate plaque growth through the phagocytosis of amyloid deposits. “Burn plaques” are often thought to reflect the plaque lysis produced by glial cells. Reactive astrocytes located in close proximity to either diffuse or compact plaques may exert a neuroprotective role in the aging brain, although the astroglial response to Aβ plaques is associated with cognitive impairment by many authors.161,163. 3) Differences in the involution of the functional glio-neuro-vascular units.
In a focal area or in a larger area spanning different regions of the brain, involutive processes can affect cellular elements in close morphofunctional relationships (neurons, glial cells and vascular structures) that configure the basic trophic and functional units to maintain the elements of the neural circuits. Vascular risk factors can result in dysregulation of the neurovascular units producing hypoxia and altered transport to the blood (including clearance of amyloid) as well as neuronal degeneration and/or neuroglial toxic responses inducing parenchymal and vascular accumulation of Aβ.181,230 The set of toxic factors produced by the elements of the neurogliovascular units leads to an acceleration of the neurodegenerative process. For this reason, it is possible to observe areas with very different alterations in cellular elements. Neurovascular unit dysfunction is a main inducer of AD.181,230,231. 4) Similarities and differences in the distinct neuroglial responses in aging, AD and other neurodegenerative diseases.
Enhanced astrogliosis and microgliosis in association with aging, AD and non-AD neurodegenerative diseases have been described. In conjunction with astrocyte dysfunction in “senescence” and dystrophy, 162 impaired microglial functionality has been demonstrated in “senescence”, asthenia, and dystrophy, affecting microglial motility, proteostasis, phagocytosis and cell signaling.3,24-26,28,162,200 The number of astrocytes has often been inversely correlated with synaptic density but not in all disorders. In AD, this inverse relationship seems to occur in all cortical brain regions, yet it was only evident in the frontal pole in association with frontal lobe degeneration. The number of astrocytes appears to be maintained throughout life, even in patients suffering from AD.24,162 In contrast, microglial density appears to increase with neurodegeneration, although the increase in microglial density associated with physiological aging is accentuated in healthy individuals over 75 years of age, exceeding that observed in individuals with AD. Moreover, microglial density seems to depend on the years of disease evolution and not on the patient’s age. 86 Indeed, it was proposed that phenotypic changes underlie most glial responses but not glial proliferation. 172
Agonal events may be responsible for atrophic/senescent subtypes of neuroglial cells, mainly due to changes in pH. 130 This possibility should be investigated in studies of human brains.
Conclusions regarding glial alterations in AD and future therapeutic perspectives
In summary, there are several aspects of neuroglia that may be particularly important to understand the pathology of AD and to develop preventive therapies.
As research progresses on the characteristics and functions of the different types of neuroglial cells, which include wide diversities of morphofunctional and gene reactions as well as involutions of various subsets of these cells (mainly focused in astroglial and microglial cells), greater new possibilities for the involvement of neuroglial cells are found both in the maintenance of brain functions in adulthood, aging and in neurodegenerative diseases, as well as in the triggering and progress of neurodegenerative processes.205,231-239 New mechanisms of neuroprotection/neuroreparation)231,233,237,238 as well as of neurodegeneration205,232-236,239 are continually being described in the scientific literature, with therapeutical possibilities (developed in the second part of this monography). As conclusions we want to highlight: 1) In the brains of individuals who suffer from AD, alterations of neuroglial cells are consistently observed in different regions of the brain, but the types of alterations observed are highly variable, both in the morphological and functional aspects, both in their neuroprotective/neuroreparative and neurotoxic nature, both in their presence in large areas and in small regions or as cells of the same subtype but with different phenotype closely intermingled. As above mentioned, it is true that there are different brain areas where important conventional astrogliosis and/or microgliosis processes can be observed (aspect that is considered in a large number of publications as a specific or characteristic “marker” of the underlying neurodegeneration in AD). However, there are also areas where there is a reduction in astrogliosis or microgliosis. In addition, as it has been shown in this monograph, there are significant differences in the relationships of neuroglial cells with amyloid plaques and altered neurons. On the other hand, insufficient research has been done on A2 and M2 anti-inflammatory cells in human AD (which may shed light on the neuropathological pathways of AD and/or define new therapeutic targets). All types of glial cells seem to be affected: astrocytes can present both astrogliosis (hypertrophy/hyperactivity and/or hyperplasia) (of A1 or A2 phenotypes) and involution (morphological and functional); microglial cells exhibit proinflammatory changes to subsets of resident and newly (?) incorporated cells as well as anti-inflammatory changes; and oligodendrocytes demonstrate a loss of cellular elements, demyelination, and a decrease in the number and function of NG2+ cells (although these cells could proliferate and try to recover the damaged neurons). All these changes can drive the manifestation of symptoms and the variable progression of AD, while many of these changes may appear in physiological senility, they are much more marked in AD. 2) Complex relationships between the various morpho-functional forms (normal, reactive, dystrophic, “senescent” – these currently valued for their expression of specific macromolecules240-242) of the main neuroglial types accompanying neurons (both normal and dystrophic) are working during all life, both in normal or abnormal circumstances (Figure 16). Neuroglia cells maintain close interrelationships (“crosstalk of glial cells")209,243 offering a modulate response of the entire neuroglial group close in contact with neurons, both normal and dystrophic, in specific areas. Neuroactive neuroglia glial substances (cytokines, chemokines, prostaglandins, NO, free radicals, …), can finally produce (directly or in-directly) neurotoxicity/neuronal involution or stimulate neuronal recovery. In the first case, the first action could tend to eliminate neurotoxic neurons to improve homeostasis, but in the case of AD can spread neurodegeneration in degenerating areas. 3) The production of amyloids, amyloid deposition in the parenchyma of nervous tissue and in the wall of blood vessels, and the accumulation of phosphorylated tau and other proteins in the soma (tangles) as well as in the axons and dendrites of neurons (dystrophic neurites) produces large and complex reactive responses in all neuroglial cells. In contrast, alterations to reactive neuroglial cells induce the formation of amyloid and aberrant intraneuronal deposits, consequently inducing morphological and functional degeneration of neurons. The neuronal or neuroglial process affected can lead to the establishment of a neurodegenerative disease; together, alterations to the cells and the affected processes contribute to disease progression. 4) The reactivity of astroglia, oligodendroglia and microglia to some extent represents mechanisms that initially seem to counteract the damaging changes in the neural milieu. These changes apparently attempt to correct neural dysfunction and neuronal circuits or eliminate the neurons that induce toxicity or that are undergoing involution/death. 5) The development of AD can be studied in experimental models from very early stages of life, especially in transgenic mice that express Ab and phosphorylated tau (which are deposited in the nerve parenchyma—amyloid—and neurons—tau protein—in the human brain). However, these studies have not provided much information regarding the pathogenesis of AD in humans, although they have enabled phenotypic variations in neurons and glial cells to be defined during the course of the disease and in aging. Many of these studies highlighted the neuropathological alterations and the differences in cognitive performance of these animals, such that the extrapolation of these results must be analyzed carefully.26,160,161. 6) Novel techniques that combine cellular and molecular approaches79-84 will reveal new insights to improve the present understanding of the genetic changes to cells that accompany the changes in neurons affected by neurodegenerative processes. Thus, it should be possible to determine what causes neurodegenerative involution and disease progression and to identify new therapeutic targets. Scheme where the complex relationships between the various morpho-functional forms (normal, reactive, dystrophic, “senescent” – these currently valued for their expression of specific macromolecules [240-242] of the main neuroglial types with the accompanied neurons (both normal and dystrophic) are considered. Neuroglia cells maintain close interrelationships (“crosstalk of glial cells”) [209, 243] offering a modulate response of the entire neuroglial group on neurons, both normal and dystrophic. Neuroactive neuroglia glial substances (cytokines, chemokines, prostaglandins, NO, free radicals, etc), can finally produce (directly or in-directly) neurotoxicity/neuronal involution or stimulate neuronal recovery. In the first case, the first action could tend to eliminate neurotoxic neurons to improve homeostasis, but in the case of AD can spread neurodegeneration in degenerating areas.

As a conclusion, it should be noted that neuroglial cells are a) fully involved in the neuropathology of AD and b) that the different types and subtypes of these cells can be both neuroprotective/neuroreparative or neurotoxic, either simultaneously or consecutively depending on the subset of cells. Future research should aim to elucidate the true role of each cell subtype and the possible transitions from their normal phenotype to a reactive state. This research should focus on each phase in the evolution of AD and develop specific strategies for each situation. Some of the specific objectives should be to determine the specific characteristics of astroglial, oligodendroglial and microglial cells from each specific region/area of normal, aged and AD brains in each phase of the disease; the neurotoxic or neuroprotective roles that these cells play; the role of each subtype in the formation of amyloid deposits (diffuse amyloid or plaques) or, conversely, in the clearance of amyloid; the effect that these cells have on the blood-brain barrier; the effects that these cells have on neuronal function; the factors that induce responses from each of these cell types; and the intracellular communication pathways that drive the phenotypic changes characteristic of each cell subtype. These issues remain largely unresolved but should be clarified through extensive research into the behavior of neuroglia.
As above mentioned in the Introduction, in the second part of this monograph (“The relationships between neuroglial alterations and neuronal changes in Alzheimer’s Disease, and the existing controversies. II Gliotherapy and multimodal AD therapy”) we will study the therapeutic possibilities that are being analyzed to try to prevent or palliate the onset or progression of this disease, based on treatments aimed at maintaining the normal functions of neuroglial cells or normalizing the alterations that occur in the progression of AD.
Footnotes
Author contributions
All the authors have been carried out jointly the research on AD and have written this monography.
Declaration of conflicting interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding:
The author(s) received no financial support for the research, authorship, and/or publication of this article.