
Abstract
Homocysteine (Hcy) is a sulfhydryl-containing amino acid, and intermediate metabolite formed in metabolising methionine (Met) to cysteine (Cys); defective Met metabolism can increase Hcy. The effect of hyperhomocysteinemia (HHcy) on human health, is well described and associated with multiple clinical conditions. HHcy is considered to be an independent risk factor for common cardiovascular and central nervous disorders, where its role in folate metabolism and choline catabolism is fundamental in many metabolic pathways. HHcy induces inflammatory responses via increasing the pro-inflammatory cytokines and downregulation of anti-inflammatory cytokines which lead to Hcy-induced cell apoptosis. Conflicting evidence indicates that the development of the homocysteine-associated cerebrovascular disease may be prevented by the maintenance of normal Hcy levels. In this review, we discuss common conditions associated with HHcy and biochemical diagnostic workup that may help in reaching diagnosis at early stages. Furthermore, future systematic studies need to prove the exact pathophysiological mechanism of HHcy at the cellular level and the effect of Hcy lowering agents on disease courses.
Introduction
Homocysteine (Hcy) is a sulfhydryl-containing amino acid, a homolog of cysteine with one additional methylene group.1,2 Hcy is not acquired through the diet, but is synthesized as an intermediate metabolite from methionine (Met) metabolism. It is converted to cysteine via the transsulfuration pathway or resynthesized back to methionine via the re-methylation pathway.3,4 In plasma, 99% of the Hcy is bound to proteins, including cysteine, and cysteinylglycine via disulfide linkages, while only 1% is found in a free reduced form. 5 For the estimation of the Hcy level, plasma tissue homogenate samples are analyzed using various methodologies such as immunoassay, capillary electrophoresis, enzymatic assay, liquid chromatography-mass spectrometry (LCMS) and high-pressure liquid chromatography (HPLC). 6 The normal Hcy levels is ranging between 5 and 15 μmol/L, while a mildly increased level is 15 to 30 μmol/L, moderate from 30 to 100 μmol/L, and a value >100 μmol/L is classified as severe hyperhomocysteinemia.7,8
The levels of Hcy can be increased through a defective metabolism of Met, due to genetic defects of the transcription of enzymes responsible for Hcy metabolism or deficiencies of cofactors involved in these pathways such as vitamins B6, B12, and folate. 9 The effect of hyperhomocysteinemia (HHcy) on human health was first described in the mid-20th century by Kilmer S. McCully. 10 Since then, many epidemiologic reports indicated that HHcy is associated with multiple clinical conditions, while controlled Hcy level in high risk group associated with improved physical and mental health.11-14 It is considered as an independent risk factor for cardiovascular disease, as well as for stroke and myocardial infarction by the American Heart Association.15,16 Although it is not directly involved in protein synthesis, the exposure to a toxic effect of Hcy, induced cytotoxicity that lead to a reduction of cultured endothelial cells viability through a direct and indirect effect on the pathway of apoptosis.17,18
Studies have identified a strong association between HHcy and induction of inflammatory determinants including the expression of adhesion molecules, leukocyte adhesion, endothelial dysfunction, oxidative stress, and reduced nitric oxide bioavailability in both human and experimental models.19,20 In HHcy state, NFκB, a transcription factor that regulates the transcription of various genes involved in inflammatory and immune responses is activated, additionally, marked increase in pro-inflammatory cytokines and downregulation of anti-inflammatory cytokines were observed. 19
In HHcy patients, supporting evidence indicate that the development of homocysteine-associated vascular disease may be prevented by the maintenance of normal Hcy levels, with conventional treatment of folate supplementation and vitamin B6 and possibly vitamin B12. 21 However, despite lowered Hcy levels, the clinical picture of pathophysiological conditions caused by an elevated Hcy level may not be reversible for certain conditions. 22
Several studies were conducted to investigate the treatment of HHcy in patients with a history of arteriosclerotic vascular disease (ASVD).23,24 A large study was conducted with more than a 1000 individuals with HHcy, to determine the effect of supplementation with vitamins B12, B6, and folic acid for 6 weeks. The study reported that there was a proportional reduction in the plasma Hcy level caused by the folic acid treatment and that the level of the reduction was higher in participants with pre-treated Hcy levels. 25 The dosage required for the treatment of HHcy may vary according to the underlying condition, however, the minimum effective dose of folic acid to obtain the maximal lowering of Hcy is 400 μg.26,27 Prolonged folic acid therapy is associated with decreased vitamin B12 blood levels and worsening symptoms of B12 deficiency, so a supplement vitamin B12 is usually taken in the form of the cyanocobalamin. 28 In a randomized controlled trial, 104 patients were divided in 2 groups, the study group received a combined nutraceutical containing 400 μg folate-6-5-methyltetrahydrofolate, 3 mg vitamin B6, 5 μg vitamin B12, 2.4 mg vitamin B2, 12.5 mg zinc and 250 mg betaine once daily for 2 months, and the control group received a supplement of folic acid (5 mg/day). The results indicated that the Hcy reduction was significantly higher in the treatment group (P < .035). 28 This may support the available evidences that vitamin supplements significantly reduce the Hcy levels in a sustained but suboptimal way, even if supraphysiological doses are used.29,30 Recent study revealed the protective role of vitamin E as antioxidant and melatonin may alleviate Hcy-induced cell apoptosis, which may add insight into therapeutic approaches to Hcy-induced damages in endothelial cells. 31
The review will discuss Hcy and its biological functions in the body, conditions that induce by or are related to HHcy, and biochemical investigations that may ease the recognition of suspected cases at early stage of disease course.
Biosynthesis and Metabolism of Homocysteine
Hcy is produced in all cells and biosynthesized from methionine through multiple steps, initiated by the demethylation of methionine (Met) as well as 3 subsequent steps.1,3 The first step is the transfer of an adenosine group from ATP to methionine by S-adenosyl methionine (SAM or AdoMet) synthetase (also called methionine adenosyltransferase, MAT), resulting in the formation of S-Adenosyl-L-methionine (AdoMet or SAM)32-34 (Figure 1).
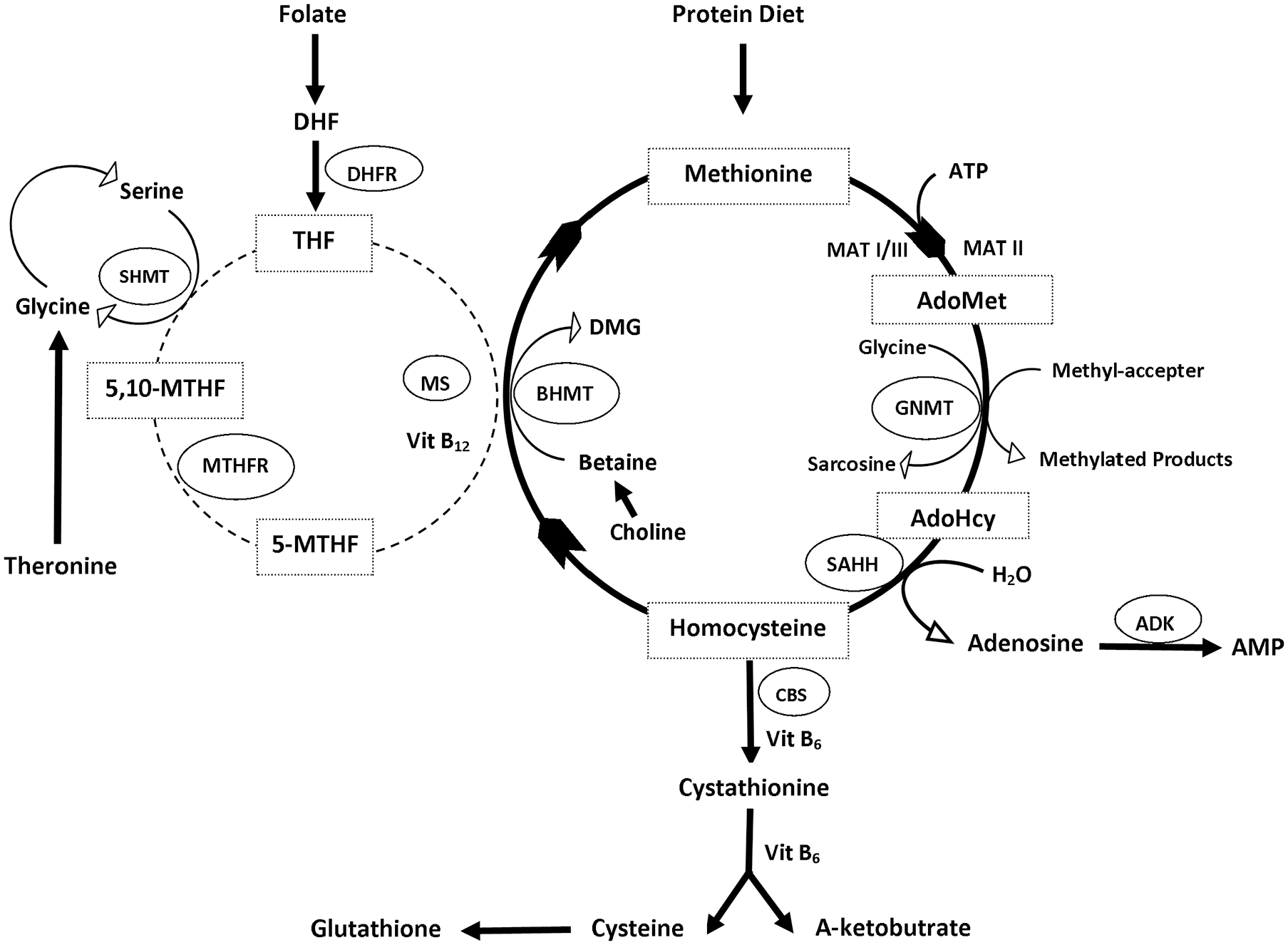
The homocysteine metabolic cycle.
In the second step, the universal methyl donor, SAM, donates a methyl group to acceptor molecules such as DNA, RNA, proteins, and neurotransmitters. 35 The resulting compound S-adenosyl homocysteine (AdoHCys or SAH), lacking the methyl group, can function as an inhibitor of most methyltransferases and is subsequently cleaved via a reversible reaction by S-adenosyl homocysteine hydrolase (SAHH) to produce adenosine and L-homocysteine.4,36,37 Finally, the L-homocysteine can be metabolized by 2 reactions, transsulfuration or re-methylation, ultimately producing L-methionine and L-cysteine, respectively.2,38
Remethylation
Remethylation involves recycling Hcy to methionine, by using vitamin B12 as a cofactor. The methionine synthase (MS) catalyzes the re-methylation reaction, restoring Met by transferring the methyl group from 5-N-methyl tetrahydrofolate (5-methyl-THF) to Hcy.39-41 In this cycle, folate is reduced to tetrahydrofolate, which is an important key player in folate metabolism as a folate acceptor molecule, which is then converted to 5,10-methylenetetrahydrofolate (5,10-methylene-THF) by the pyridoxal phosphate (PLP)-dependent serine hydroxymethyltransferase (SHMT). 42 Methylenetetrahydrofolate reductase (MTHFR) reduces 5,10-methylene-THF to 5-methyl-THF. 43
There is another pathway of remethylating Hcy that uses an enzyme called betaine-homocysteine methyltransferase (BHMT). The betaine pathway is restricted to the liver and kidney where betaine can serve as methyl donor molecules. In this reaction, the methyl group is transferred from betaine to Hcy to produce methionine and dimethylglycine (DMG).41,44,45 Here choline plays a significant role in Met regeneration, as it is oxidized to betaine, which can be used in this conversion of Hcy.46,47
Transsulfuration
In the transsulfuration process, Hcy is irreversibly converted to cysteine by cystathionine b-synthase (CBS), which is followed by the catalysis done by cystathionine c-lyase (CTL). Both enzymes need the cofactor pyridoxal-50-phosphate (vitamin B6) to function. 48 Serine can be enzymatically added to homocysteine by CBS and vitamin B6, to form cystathionine, which can be cleaved by CTL to form cysteine. 49 Once cysteine is formed, it can be used in protein synthesis and glutathione (GSH) production and cannot be converted to back to Hcy. 50
Causes of Hyperhomocysteinemia
Enzyme defects associated with Hcy metabolism are considered the most prevalent cause of HHcy. The enzyme defects has been researched, especially the polymorphisms of the main enzymes involved in Hcy metabolism such as Cystathionine b-synthase (CBS) deficiency, Methylenetetrahydrofolate reductase (MTHFR) deficiency, Methionine synthase deficiency, and Methionine adenosyltransferase deficiency (Table 1).37,51,52 In addition to genetic causes, many other factors related to age, lifestyle such as cigarette smoking, alcohol consumption and nutritional deficiencies in folic acid, vitamin B6, vitamin B12, and betaine are as responsible for HHcy.53-56 In this review, we will discuss the main enzymatic defects in this pathway.
Causes of hyperhomocysteinemia.

Cystathionine beta-synthase (CBS) deficiency or classical homocystinuria
Classical homocystinuria (HCU) (OMIM 236200), is an autosomal recessive disease caused by biallelic pathogenic variations in the CBS gene. 57 Deficiency of the CBS enzyme causes elevated tissue and plasma levels of Hcy and its precursor, methionine. 58 Typically, patients can manifest a wide range of symptoms with variable severity involving the ocular, skeletal, vascular, and central nervous systems.59,60
The prevalence of CBS deficiency has been reported as 1:200 000 to 1:335 000 and >200 pathogenic variants have been described in the CBS gene, however, mutations such as p.Ile278Thr, p.Thr191Met, p.Gly307Ser, and p.Trp323Ter are the most prevalent mutations and represent half of all HCU alleles.61-63 Considering the significant effect of genetic polymorphisms on the increase of the HCys level, current studies are investigating the correlation between the polymorphisms and stroke events, however, the results are still conflicting. 56 Not all polymorphisms in CBS have an effect on enzyme activity, however a T833C polymorphism in CBS, caused mild HHcy in different ethnic groups.64-66 Current treatment options for CBS are very limited and often inefficient, partially due to low patient compliance with a very strict dietary regimen. 67 However recent studies shows the efficacy of some novel therapies including enzyme replacement and gene therapy approaches. 68
Methylenetetrahydrofolate reductase (MTHFR)
Methylenetetrahydrofolate reductase (MTHFR) deficiency (OMIM 236250), an autosomal-recessive inheritance disease, is caused by a mutation in the MTHFR gene which encode MTHFR, a key enzyme of folate metabolism in the process of one-carbon metabolism. 69 Polymorphisms of MTHFR would cause impaired methylation as well as a deficiency of folate, and a wide range of diseases including cardiovascular, tumors, neurologic, and psychiatric disorders.70,71 One of the most studied polymorphisms in MTHFR is C677T.72-74 This polymorphism is responsible for an increase of Hcy concentration and folate deficiency compared to a normal genotype individual. 75 It is estimated that 10% of the global population is homozygous (TT genotype), which may vary in different populations reaching 25%. 42 The treatment in MTHFR is symptomatic including the treatment of associated neurological symptoms. 76 Vitamin supplementation should be considered in these patients including vitamin B12, folic acid, vitamin B6, betaine, and methionine. 77
Methionine synthase
Methionine synthase promotes the methyl group transfer from methylated folate to homocysteine to yield methionine, and Cbl act as a cofactor in this catalytic reaction. This enzyme is encoded by the MTR gene. Mutations in this gene are the underlying cause of methylcobalamin deficiency cblG-type.78,79 In patients with a methionine synthase deficiency (CbIG), complementation studies on cultured fibroblasts indicated a cblG defect. 80 This deficiency is rarely reported in literature, and the patients do not have specific neurological symptoms for example, blindness or leukoencephalopathy associated with normal vitamin B12 and folate, hyperhomocysteinemia with hypomethioninemia in the absence of methylmalonic acid. 74
Acquired and inherited disorders of cobalamin
Cobalamin (Cbl-Vitamin B12) is an essential cofactor for MS in the folate cycle to ultimately produce the 5-Methyl THF which provides a methyl group to convert homocysteine to methionine.69,81 Inborn errors of cobalamin metabolism can affect its absorption (intrinsic factor deficiency, Imerslund-Gräsbeck syndrome), transportation (transcobalamin deficiency), as well as genetic defects of the intracellular cobalamin metabolism such as CblC, CblD, CblE, CblF, and CblG.82-84 Megaloblastic anemia, pancytopenia and failure to thrive are the main manifestation of Cbl deficiency. However, if the diagnosis is delayed, it may be accompanied by irreversible neurological deficits. 83 Cbl deficiency rarely requires instant therapy, however, treatment should be started timeously to prevent severe neurologic symptoms (eg, seizures, gait disturbances, mental changes and extensive sensory defects) due to the risk of irreversibility. 85
Methylation disorders
Inherited methylation disorders are a group of disorders affecting the transmethylation processes in the metabolic pathway between methionine and Homocysteine, including methionine adenosyltransferase I/III, glycine N-methyltransferase, Sadenosylhomo-cysteine hydrolase and adenosine kinase deficiencies. 86 Although isolated hypermethioninemia is the biochemical hallmark of this group of disorders, mild to moderate HHcy can be present in all patients. 87 Three of these directly affect the reactions in the methionine pathway. The first is the conversion of methionine to AdoMet, catalyzed by methionine adenosyltransferase. The second enzyme is glycine N-methyltransferase (GNMT), which transfers a methyl group to glycine producing sarcosine. The third enzyme, S-adenosylhomocysteine hydrolase (SAHH), is a homotetrameric enzyme, which converts S adenosylhomocysteine to homocysteine and adenosine.88-90 In methylation disorders, a low methionine diet can be beneficial in patients with MAT I/III deficiency, and to a lesser extend in SAHH and AKD. S-adenosylmethionine supplementation may specifically be useful in patients with MAT I/III deficiency. 86
Conditions Associated With Hyperhomocysteine
Hcy function in folate metabolism and choline catabolism is fundamental for the synthesis of different sulfur-containing amino acids and methylated compounds which are important for may cellular pathways.42,91 Numerous studies demonstrated that the disruption of the Hcy metabolism which lead to HHcy by common MTHFR gene polymorphism, increases the risk for several complex disorders and it plays an important role in the pathogenicity of such disorders, including the cardiovascular system for example, congestive heart disease and arthrosclerosis.92-94 In the central nervous system, the disorders include cognitive impairment, Parkinson’s disease, Alzheimer’s disease (AD), multiple sclerosis and epilepsy. 95 In addition, an elevated Hcy was linked to osteoporosis, chronic renal failure, hypothyroidism, insulin resistant diabetes, polycystic ovarian syndrome, and gastrointestinal disorders.96-100 Although the molecular mechanism in this role has not been fully defined, age related disorders such as acoustic dysfunction, and age related macular degeneration has been reported.101-104
Stroke and cardiovascular diseases
Literature support the theory of a correlation between HHcy and the risk for peripheral vascular diseases, including stroke, venous thromboembolism and cardiovascular disease, for example congestive cardiomyopathy, myocardial infarction and coronary artery disease. 105 A potential mechanism is the thrombotic activity of Hcy and its direct effect of endothelial dysfunction, where the Hcy acts as an inhibitor of endothelial nitric oxide synthase (eNOS), which will cause reduced bioavailability of NO, through the inhibitory effect of asymmetric dimethylarginine (ADMA). 106 Findings from a clinical study investigating patients with heart failure, support through preclinical evidence that the myocardium is especially vulnerable to damage by HHcy, which is associated with the production of reactive oxygen species and cause the progression of cardiovascular disease and left ventricle remodeling.107,108
Cognitive impairment, Alzheimer’s disease, Parkinson’s disease and epilepsy
Several case control studies confirmed a positive correlation between HHcy as a neurotoxic condition and both vascular dementia and AD. However, it remains unclear whether an elevated blood homocysteine level is a direct risk factor for AD or possibly, poor vitamin nutrition in the elderly. 101 The mechanism through which high levels of Hcy cause AD is still being investigated, however, in an experimental study, HHcy resulted in increased gene expression of proinflammatory markers such as IL1b and TNFa in microglia and an increased expression of kinases in neuronal cells. 109 Studies have also shown an increase in neurodegeneration due to homocysteine-related oxidative stress, causing an increase in the production of superoxide and other reactive oxygen species, and apoptosis. 110 Another proposed mechanism for Hcy neurodegeneration involves its role as an agonist for AMPA (both metabotropic and ionotropic) and NMDA receptors. All those changes in vascular smooth muscle cells, provide further neurotoxic peculiarity to HHcy as a risk factor for neurodegenerative diseases. 111 Recently, an experimental model, linked the HHcy to increased oxidative stress, upregulated expression of proteins that promote blood coagulation, exacerbated blood-brain barrier dysfunction and promoted the infiltration of inflammatory cells into the cortex in traumatic brain injury (TBI). 112
Gastrointestinal disorders
There is growing evidence that HHcy is associated with inflammatory bowel disease (IBD) and many autoimmune diseases.113,114 In a meta-analysis of 28 studies, the Hcy levels were significantly higher in IBD patients, compared to the controls. 115 The pathophysiological mechanisms leading to vascular damage in hyperhomocysteinemia are multifactorial, and still poorly understood. Studies have also shown that the colonic mucosa of patients with IBD has a higher level of Homocysteine and it has been hypothesized that the lamina propria mononuclear cells (LPMC) play an important role in homocysteine production. 116
Chronic renal diseases
Hcy has been documented in patients with chronic renal failure (CRF), on dialysis or after a kidney transplant, at higher concentrations than in individuals without kidney disease. 117 A study was conducted with 89 renal failure patients on dialysis, to determine the frequency of the MTHFR gene mutation or polymorphism and hyperhomocysteinemia. The study confirmed the high prevalence of hyperhomocysteinemia in patients on dialysis, diagnosed in 76 patients (85.39%), as well as the high incidence of the C677T and A1298C mutation, in 42 (47.19%) and 29 (32.58%) patients, respectively. 118
A Clinical Approach for Hyperhomocysteinemia
It must be noted that HHCY, the biochemical hallmark of a large group of diseases that characterized by variable presentation affecting many organs, however, the predominant associated features are hematological and unexplained neurological signs and symptoms. 22 Predominantly, HHcy is associated with vitamin B12 deficiency, where the measurements of metabolites, such as methylmalonic acid (MMA) and Hcy, are more sensitive in the diagnosis than the measurement of serum B12 levels alone, with 98.4% with elevated serum MMA levels, and 95.9% with elevated serum homocysteine levels in B12 deficiency cases.119-121 The differing treatments for each genetic cause of HHcy necessitate identifying a specific underlying causes, in order to provide paradigmatic treatment. In suspected cases, a careful clinical evaluation that using a variety of metabolites including, total Hcy, PAA, Vitamin B6, Vitamin B12 levels, and serum and urine levels of MMA rather than just Hcy will help elucidate the cause of HHcy, should facilitate their exclusion (Figure 2). The level of elevation and Hcy and the status of associated metabolites will help in narrowing the diagnosis. Combined sever HHcy with high methionine in PAA frequently seen in CBS deficiency (classical homocystinuria), while in methylation disorders (MAT I/III, GNMT, ADK, and SAHH), HHcy is moderately elevated. In case if HHcy associated with low methionine, MTHFR and MS should be considered. The elevation of MMA is also important if associated with HHcy because it is indicate inherited cobalamin disorders or vitamin B12. After accurate interpretation of these metabolites the diagnosis can be confirmed by investigations at the levels of metabolites, enzymatic studies and/or molecular genetic analysis.86,122

Algorithm for diagnosis of hyperhomocysteinemia.
In several countries, C3-propionylcarnitine and methionine is used as markers in newborn screening programs in asymptomatic newborns, where C3-propionylcarnitine is used as a marker to detect patients with vitamin B12 deficiency, intracellular Cbl disorders, while elevated methionine used for CBS deficiency and methylation defects, such as MAT I/III and GNMT. 90 After making a diagnosis and initiating a treatment plan, follow-up is important to determine the patient’s response to therapy. In mild vitamin B12 deficiency, depending on the underlying cause, frequent measurements of serum vitamin B12, Hcy, and MAA levels is recommended for monitoring of therapy.123-126
Conclusion
Hcy is now considered as a risk marker for cardiovascular and cerebrovascular disease in addition to other modified and non-modified individual factors. Simultaneous measurement of vitamin B12, Hcy, MMA, and PAA is accepted as a sensitive method of screening for several conditions associated with HHcy. HHcy-induced inflammation could play a role in blood brain barrier (BBB) dysfunction and the pathogenesis. Thus, the elimination of excess homocysteine could be a potential therapeutic intervention therefor may be value in preventative supplementation, especially folic acid, vitamin B12 and betaine, if the foods indicated are not being consumed in sufficient quantities.
Footnotes
Funding:
The author received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests:
The author declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ contributions
FAM prepared and summarized the literature, designed the figures and tables and wrote the manuscript.
Availability of data and materials
All data generated or analyzed during this study are included in this published article, any additional data/files may be obtained from the corresponding author.