
Abstract
Sepsis is a systemic inflammatory disease resulting from an infection. This disorder affects 750 000 people annually in the United States and has a 62% rehospitalization rate. Septic symptoms range from typical flu-like symptoms (eg, headache, fever) to a multifactorial syndrome known as sepsis-associated encephalopathy (SAE). Patients with SAE exhibit an acute altered mental status and often have higher mortality and morbidity. In addition, many sepsis survivors are also burdened with long-term cognitive impairment. The mechanisms through which sepsis initiates SAE and promotes long-term cognitive impairment in septic survivors are poorly understood. Due to its unique role as an interface between the brain and the periphery, numerous studies support a regulatory role for the blood-brain barrier (BBB) in the progression of acute and chronic brain dysfunction. In this review, we discuss the current body of literature which supports the BBB as a nexus which integrates signals from the brain and the periphery in sepsis. We highlight key insights on the mechanisms that contribute to the BBB’s role in sepsis which include neuroinflammation, increased barrier permeability, immune cell infiltration, mitochondrial dysfunction, and a potential barrier role for tissue non-specific alkaline phosphatase (TNAP). Finally, we address current drug treatments (eg, antimicrobials and intravenous immunoglobulins) for sepsis and their potential outcomes on brain function. A comprehensive understanding of these mechanisms may enable clinicians to target specific aspects of BBB function as a therapeutic tool to limit long-term cognitive impairment in sepsis survivors.
Keywords
Introduction
Sepsis is a debilitating systemic inflammatory process involving multiple organ systems that is preceded by an infection. It is the 10th leading cause of death in the United States with an annual financial burden for patients and survivors that exceeds $20 billion. 1 Through mechanisms that remain largely poorly understood, sepsis can induce acute and chronic changes in the central nervous system (CNS), particularly at the blood-brain barrier (BBB). A compromised CNS can lead to sepsis-associated encephalopathy (SAE), a well-characterized state of cognitive impairment and neurological dysfunction often seen in the acute phase of sepsis. Multiple pathways have been investigated for their contribution to the sepsis-associated compromise of the BBB.2,3 This review integrates current clinical knowledge of sepsis with mechanistic insights from both clinical studies and preclinical animal models of sepsis. The overall goal of this review is to understand how sepsis pathophysiology perturbs the integral functions of the cells and proteins that comprise the BBB. Thus, we provide insights to uncover how a compromised BBB may lead to SAE or permanent brain dysfunction in sepsis survivors.
Sepsis Pathophysiology
Clinical sepsis presentation
The current definition from the Sepsis-3 consortium describes sepsis as a life-threatening organ dysfunction caused by a dysregulated host response to an infection. 4 The most common precipitating sites for sepsis are the respiratory system, genitourinary system, and the abdomen. Sepsis may present as a combination of various non-descript signs and symptoms making early diagnosis difficult. For example, patients may present with fever, cold, pain, delirium, increased heart rate, shortness of breath, diarrhea, and/or low blood pressure. The diagnosis and management of sepsis has changed dramatically over 30 years. The historical definition of sepsis was focused primarily on inflammation and incorrectly portrayed sepsis as a sequential process that eventually ends in septic shock (Figure 1A). In 2016, Sepsis-2 criteria were revised to current Sespis-3 criteria to improve consistency in classification in epidemiological and clinical trials. The revised Sepsis-3 classification shown in Figure 1B focuses on accelerated recognition and management of sepsis. 5

Comparison of the past and present guidelines for diagnosis of sepsis and septic shock. (A) Past guidelines stressed that the diagnosis and progression of sepsis from a systemic inflammatory response to multiple organ dysfunction was sequential rather than multifactorial. (B) The current guidelines for defining sepsis and septic shock stress that multiple linked considerations are necessary for an accurate diagnosis. Both the past and present guidelines involve a clinical screening tool for patients likely to have sepsis that includes a clinical characterization of the severity of the disease. Clinicians have traditionally used a Sequential Organ Failure Assessment (SOFA) system to categorize the severity of organ dysfunction in sepsis, which associates a higher SOFA score with increased mortality.4,6 New sepsis guidelines employ a quick Sequential Organ Failure Assessment (qSOFA) score, which is a modified version of the SOFA score that includes altered mental status, a systolic blood pressure ⩽ 100 mm Hg, and a respiratory rate ⩾ 22/min. Patients with a qSOFA score ⩾ 2 have an overall in-hospital mortality risk greater than 10%.4,7 If warranted, further clinical analysis can be completed using other SOFA criteria. 4
In recent years, the focus in clinical treatment has shifted to severe sepsis and septic shock, which has increased survival in hospitalized patients diagnosed with severe sepsis and critically ill patients with septic shock who have a higher risk of multi-organ failure complications and death. 8 This heterogeneous presentation of clinical sepsis makes disease management and appropriate therapeutic interventions difficult. Some challenges associated with the management of sepsis include late diagnoses, poor prognoses, inadequate therapeutics, and post-sepsis complications. These challenges stem from late recognition and difficulties associated with differentiation of sepsis from other illnesses in its early stage. In the later stages of sepsis, recognition becomes easier, yet sepsis is more difficult to treat and often coincides with multi-organ failure.5,8,9
Most sepsis cases are hospital acquired and are often comorbid with prior injury, such as stroke, trauma, or post-surgery. Most cases of hospital-acquired sepsis are treated in the intensive care unit (ICU). However, ICU heterogeneity can make sepsis more common in one ICU versus another. For example, there is a higher incidence of sepsis in a trauma ICU as opposed to a surgical ICU. 8 Alternatively, a significant number of sepsis patients are admitted to the hospital or directly to the ICU via the Emergency Department (ED); most of these patients present with community-acquired sepsis from pneumonia or complications from other comorbid conditions such as diabetes.10,11 In addition to the patient setting, the development of sepsis often depends on several risk factors, such as age, where a proportionate relationship exists between increasing age and sepsis acquisition.12,13 Male sex, non-white ethnicity, and preexisting conditions such as Alzheimer disease (AD), HIV, or cancer are also risk factors for acquisition.8,13–15
Current experimental animal models of sepsis
Understanding the mechanisms involved in the pathophysiology of sepsis requires the use of animal models that adequately reproduce several features of the human disorder including both inflammation and infection. The most common animal models are cecal ligation and puncture (CLP), the colon ascendens stent peritonitis (CASP) model, endotoxin injection, and bacterial infusion. 16 The CLP model is regarded as the gold standard for human-like sepsis progression in animal models. 17 Execution of this model necessitates leakage of polymicrobial feces into the peritoneum after the cecum is punctured with a needle. Disease severity is modeled by controlling needle size and number of punctures; however, a major limitation of this model is the failure to maintain continuous fecal leakage due to abscess formation and necrosis of the punctured bowel. Some investigators also administer antibiotics either at the time of injury or at intervals post injury. Although antibiotic administration is an additional feature which mimics the treatment regimen in human patients, the use of different antibiotic classes and dosing paradigms across laboratories may confound the interpretation of findings when results are compared between laboratories.16,18 The CASP model is a newer model recently introduced to counter the flaws of the CLP model.16,19 The model involves the insertion of a stent at the ascending portion of the colon, allowing continual leakage of feces into the peritoneum.16,20 Despite resemblance to human-like sepsis progression, the drawbacks to this model include animal variation in colon size, fecal content, and the volume of feces that leaks into the peritoneum.16,21 Although these two models have provided remarkable insights to understand the pathophysiology of sepsis in humans, they fail to fully recapitulate the comprehensive clinical progression of sepsis in humans. 16
Two alternative sepsis models involve injection or infusion of endotoxin or bacteria. The endotoxin model typically involves injection of lipopolysaccharide (LPS) endotoxin, a component of Gram-negative bacterial cell walls which signals most commonly through toll-like receptor-4 (TLR4). Administration of LPS via different routes (ie, intraperitoneal, intravenous, or intracerebroventricular) initiates a cytokine storm that results in the release of tumor necrosis factor alpha (TNF-α) and numerous interleukins (ILs; IL-1, IL-6, and IL-10). Injection of LPS mimics many classical signs and symptoms of sepsis-induced inflammation, thereby providing a basic understanding of how inflammation activates the immune response in sepsis.16,22 One major limitation of the LPS model is the lack of integration of the infection component. A second limitation is that very large endotoxin doses are required in many rodent models to mimic the pathological profile of the clinical sepsis picture observed in humans. 23 Bacterial injection is a less widely used model involving infusion of a bacterium, usually Escherichia coli or Staphylococcus aureus, to initiate both inflammation and infection.16,24,25 Different bacterial strains used for infection present a challenge in this model, as they will produce different patterns of sepsis progression. 26 Thus, the characteristics of the sepsis model must be considered when interpreting the effects of sepsis on the CNS and other organ systems.
The CNS in sepsis: sickness behavior and SAE
A critical role for the CNS in the pathophysiology of sepsis has emerged over the past 2 decades. Several recent reviews address this topic in excellent detail.13,27–30 One important contribution of the CNS is “sickness behavior.” Sickness behavior is a response seen in sepsis characterized by fever, adaptive behavioral changes, and neuroimmune changes. 31 The response is governed primarily by systemic interactions with the vagus nerve (VN) and circumventricular organs (CVOs). The VN is an important mediator of inflammation. Septic mice that underwent a vagotomy (VGX) surgery exhibited an increase in the synthesis of inflammatory cytokines compared with sepsis-only mice.27,32–34 In contrast, stimulation of the VN in septic animals resulted in an overall reduction in the synthesis of inflammatory cytokines, leukocyte recruitment, and endothelial activation.34–36 The VN also relays peripheral information to the medullary autonomic nuclei, whereas the CVOs may serve as sensors for inflammatory mediators, primarily cytokines, and serve as the foci for neuroimmune communication between the peripheral circulation into the brain parenchyma. Many of these neuroimmune communication circuits are well described, but the underlying mechanisms that regulate these pathways remain poorly understood.37,38 For example, activation of the nucleus tractus solitarii and locus coeruleus by inflammatory mediators subsequently activates autonomic nuclei, behavioral, and neuroendocrine centers.39,40 The summative effect can be observed as depression, social withdrawal, increased heart rate, poor blood pressure control, or altered vigilance. 18
In addition to sickness behavior, patients with acute sepsis may have changes in brain function that present as delirium, seizures, psychological disorders, abnormal motor movements, and increased mortality.39,41 Changes in brain function are most commonly manifested as delirium. Whereas sepsis-associated delirium usually presents as decreased activity, a hyperactive form associated with agitation may be seen in some patients. 39 Tools that can be used to confirm sepsis-associated delirium include medical history, blood chemistry, electrolyte balance, the ICU screening checklist, Confusion Assessment Method, and Glasgow Coma Scale.39,42 Sickness behavior and/or delirium may progress to a more severe phenotype, SAE, which is regarded as a diagnosis of exclusion. 43 It is characterized by impaired consciousness, seizures, delirium, coma, focal cognitive deficits, and alterations in electroencephalogram (EEG) patterns. 44 Patients with SAE have increased mortality, long-term neurological decline, memory lapse, inattentiveness, disorientation, and verbal difficulties. 45
Alterations in EEG wave patterns often predict SAE outcome, and EEG reactivity is associated with mortality even at 1 year post severe sepsis.44,46 For example, a recent study showed resting-state EEG changes in sepsis survivors at 6 to 24 months after hospital discharge, including increased delta and sigma activity compared with control patients. 47 Changes in EEG frequencies can be associated with changes in brain function. For example, slowing alpha activity with increased theta activity reflects cortical dysfunction and can occur in patients with mild to moderate encephalopathy. Slowing of delta activity is often associated with more severe neurocognitive decline and indicates impaired function in deeper brain structures, such as the basal ganglia. 44 Whereas the evaluation of EEG can be sensitive to SAE diagnosis in the absence of neurological examination abnormalities, it has poor specificity and can be hampered by sedation and analgesia.45,48
Ischemia is another common complication of early sepsis due to drastic changes in systemic blood pressure. 41 A number of human clinical studies support the premise of decreased cerebral blood flow (CBF) in acute sepsis.49–52 The abrupt change in blood pressure with added sepsis-associated coagulopathy causes reduced blood flow to neurons. The hippocampal region and watershed areas are affected more often than other brain regions when this occurs. Autopsies in patients who died from septic shock revealed consistent ischemic and hypoxic insults in areas particularly susceptible to low blood flow (eg, amygdala, frontal junctional cortex, etc) and in autonomic centers. 53 Furthermore, the autopsies of 7 delirious ICU patients in another study revealed pathological lesions in the hippocampus, striatum, and pons triggered by ischemia or hypoxia. 54 The presence of ischemia in post-mortem studies strongly suggest that vascular irregularities and alterations of CBF occur during sepsis. Importantly, multiple clinical observations support the concept that, in the absence of cerebrovascular occlusion (ie, stroke), impaired cerebral autoregulation and hypotension may be the primary drivers of tissue hypoxia and cerebral ischemia observed in sepsis patients.55–57
A recent study in rats conducted by Towner et al 58 showed that CBF in the thalamus and cortex is significantly increased 24 hours post LPS injection but significantly reduced 6 weeks post LPS injection when compared with saline controls. Preceding human clinical studies also support the decrease in CBF at 24 hours observed by Towner and colleagues, yet there remains a paucity of literature on how sepsis may affect long-term CBF in human patients. Overall, coincident alterations in cerebral blood and cardiovascular collapse in sepsis emphasize the importance of fluid resuscitation as a crucial component of sepsis management. The ideal type of fluid (colloid vs crystalloids) and ideal composition used to treat septic patients remains controversial. A total of 3 clinical trials demonstrated that colloid use in sepsis treatment failed to show a clear benefit.59–61 In addition to the findings from this study, the restricted accessibility, safety issues, and the expensive value of colloids shifted the debate toward identifying the ideal crystalloid composition (eg, Ringer lactate, Ringer acetate, etc). 62 We refer the reader to excellent reviews regarding optimal fluid therapeutic strategies in sepsis.62–64
The utilization of magnetic resonance imaging (MRI) in the diagnosis of SAE offers a unique opportunity in capturing some of the morphologic, ischemic, and metabolic alterations associated with sepsis. A summary of MRI findings in acute sepsis is shown in Table 1. In particular, diffusion-weighted imaging (DWI) and the apparent diffusion coefficient (ADC) are 2 MRI modalities currently used in assessing BBB breakdown caused by vasogenic (extracellular) or cytotoxic (intracellular) edema. 73 Cytotoxic edema typically caused by ischemia, hypoxia, or vasogenic edema is the most consistently reported MRI change associated with SAE.73–76 Early detection of BBB breakdown by gadolinium (Gd) could establish an adequate therapeutic window for current and future septic treatments, but human studies are limited.77–79 A recent study in rats revealed a significant increase in the infiltration of Gd in the cortex, hippocampus, and thalamus 24 hours and 1 week post LPS injection. 58 Gd use may also cause a substantial risk of nephrogenic systemic fibrosis, a risk factor which suggests that Gd-based imaging in the CNS should be evaluated on a case-by-case basis.
MRI imaging studies in patients with sepsis.

DWI, diffusion-weighted imaging; MRI, magnetic resonance imaging.
Sedatives are often administered in the ICU when treating sepsis. A study conducted by Qiao et al 80 in rats showed that the application of dexmedetomidine and midazolam improved survival and reduced cytokine levels and splenic apoptosis in septic mice. Another systematic review by Zamani et al 81 emphasized the importance of the kind of sedative used in treating sepsis; the findings from this study revealed that dexmedetomidine improved short-term mortality when compared with other sedatives. It is widely thought that the neuroprotective effects of dexmedetomidine result from neuronal death prevention, suppression of inflammatory cytokines, and modulation of neurotransmitters released in the sympathetic nervous system.81–84 However, a limitation noted by Zamani et al was the small sample size included in the clinical studies. It is important to note that the tools used in the confirmation of sepsis-associated delirium are not helpful in ICU-sedated patients who may otherwise exhibit signs of delirium. 43
Sepsis also affects long-term neurological outcomes. The greatest risk factor for long-term impairment is the duration of delirium in the acute phase of sepsis and the increased ventricle-to-brain ratio as calculated by MRI.39,41,85,86 A seminal study published by Iwashyna et al 87 suggested that up to 70% of sepsis survivors may exhibit lasting neurological impairment, including alterations in mood, cognition, and motor function. Cognitive, motor, and mood impairments are three of the most common long-term neurological outcomes in septic patients. 41 Current evidence also suggests an increased susceptibility to other neurodegenerative disorders such as stroke or AD post sepsis insult. 88 Thus, patient populations that are the most vulnerable to long-term neurologic decline post sepsis are the elderly and patients with preexisting neurodegenerative diseases.12,13,39,88 The consequences of sepsis on both acute and chronic neurological outcomes demonstrate a critical need to understand the mechanisms involved in SAE development. Harnessing this knowledge will provide essential therapeutic avenues to limit SAE progression and protect against long-term neurological impairment or dysfunction. The remainder of this review will focus on the role of the BBB in sepsis-associated cognitive dysfunction, as preclinical and clinical investigations have uncovered 3 primary BBB-linked mechanisms that contribute to the cause of SAE and the associated short-term and long-term cognitive dysfunction: (1) activation of neuroinflammation, (2) microcirculatory dysfunction, and (3) increased neuronal excitotoxicity.39,89
Mechanisms of BBB Dysfunction in Sepsis
BBB overview
This section will provide a brief overview of the cell biology and physiology of the BBB, as the focus of this review is the BBB in sepsis. BBB and other associated cell types within the neurovascular unit that are affected by sepsis are shown in Table 2.39,107 The BBB is a highly selective, dynamic, and semipermeable biological interface between the brain parenchyma and cerebral circulation. Preservation of BBB integrity protects normal brain function and is dependent on maintaining a precise cerebral homeostasis driven, in large part, by ion and gas concentrations and nutrient availability. The BBB’s unique structure is composed of endothelial cells, astrocytes, pericytes, and a basal lamina. The endothelial cells are joined by tight junctions (TJs), and surrounding pericytes and astrocytes, which associate with the basal lamina to protect the brain parenchyma. Endothelial cells are sealed or joined together by TJs which are composed of occludin, claudin, and cadherin proteins. Because paracellular movement of compounds around and between endothelial cells is highly restricted, active transport is required to move polar solutes and nutrients across the BBB into the brain parenchyma. 108 However, water, small gases, and small- to moderate-sized lipid-soluble compounds can enter the brain passively.109,110 Primary efflux transporters include permeability glycoprotein (P-gp) and breast cancer resistance protein (BCRP), which actively pump compounds out of the brain and back into circulation. 108 Often, these transporters further restrict the permeability of compounds and drugs which may otherwise have the molecular characteristics to passively cross the BBB. 111 Readers are referred to many excellent reviews on the BBB for a more in-depth discussion of these and other topics pertaining to the BBB.37,88,109,112,113
BBB cell types affected by sepsis and consequential outcomes.
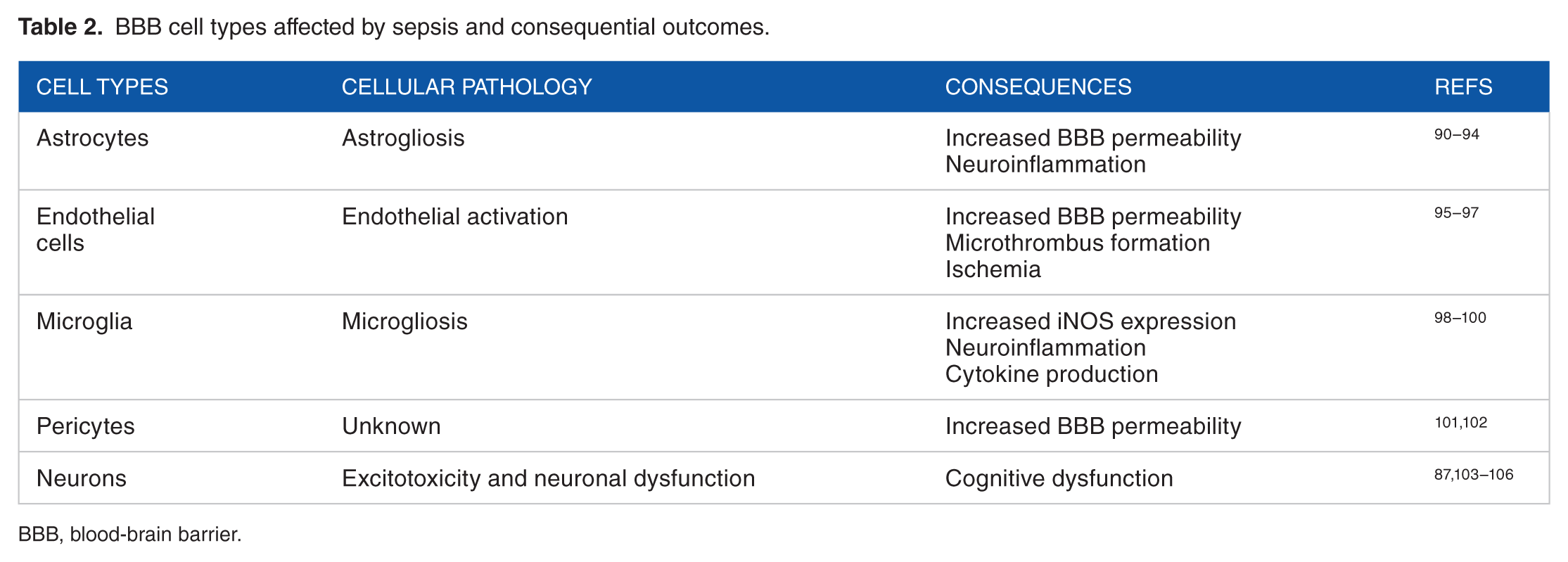
BBB, blood-brain barrier.
Neuroinflammation and BBB permeability
A probable starting point of sepsis-induced acute brain dysfunction is the initiation of neuroinflammation, but the mechanism by which this occurs is not well understood. 51 Neuroinflammation is a response to CNS disruption or dysfunction and is typically found in all neurological disorders. 114 Current literature suggests that neuroinflammation in sepsis begins when immune cells recognize foreign pathogen-associated molecular patterns (PAMPs) such as LPS, flagellin, fimbriae, peptidoglycan, heat shock proteins, and DNA fragments, which are encoded as “danger signals” to the host. Recognition of PAMPs causes the release of proinflammatory cytokines in the periphery. 115 Inflammatory mediators may enter the brain by numerous mechanisms that include transcellular diffusion, solute carrier proteins, receptor-mediated transcytosis, and adsorptive transcytosis. 37 Many cytokines enter the brain through receptor-mediated endocytosis on brain endothelial cells. For example, during inflammation, TNF-α is upregulated and its transportation from blood to brain parenchyma is increased, primarily through receptor-mediated endocytosis of its receptors, tumor necrosis factor receptor 1 (TNFR1) and tumor necrosis factor receptor 2 (TNFR2).116,117 Molecules originating in peripheral or CNS tissues may activate vascular endothelium and various leukocytes to produce hormones that facilitate their entry into the brain. For example, Nishijima et al 118 observed that prostaglandin E2 enhanced transport of serum-insulin-like growth factor 1 across the BBB.
Cytokine production contributes to neuronal dysfunction in sepsis in addition to many other neurological disorders. Cytokine infiltration enhances the activation of endothelial cells and microglia, which ultimately leads to loss of neuronal function. Activation of the endothelium leads to enhanced activity of the coagulation cascade, microthrombus formation, and ischemia, which, in turn, promotes increased BBB permeability and leukocyte infiltration. This process triggers neuronal damage, apoptosis, and brain edema.119,120 Cytokine-mediated microglial activation occurs simultaneously with endothelial cell activation. Although the normal microglial response is to phagocytose-injured neuronal cells and clear debris, sustained and dysregulated microglial activation is highly detrimental to specific regions of the CNS. Thus, persistent microglial activation enhances the production of inflammatory cytokines and reactive oxygen species (ROS), which perpetuates a vicious cycle of increased BBB permeability coupled with neuronal damage and apoptosis.116,119 Collectively, neuronal apoptosis and microglial activation are 2 primary mechanisms that increase the activity of inducible nitric oxide synthase (iNOS) activity and generation of nitric oxide (NO). Neuronal apoptosis is further exacerbated due to neuronal sensitivity from increased levels of NO produced by activated microglia.121,122 Intriguingly, iNOS levels are elevated in sepsis and are highest in deceased septic patients.70,100 This increased iNOS activity could also be responsible for the cardiovascular collapse seen in sepsis.53,100,117 It is likely that this cardiovascular collapse also affects the cerebral microcirculation and leads to subsequent sepsis-associated brain dysfunction.
Sepsis, particularly Gram-negative sepsis, has been shown to upregulate caveolin-1 at the endothelial membrane. 123 Increased caveolin-1 has recently been shown to increase the amount of peripheral immune infiltration into the brain. 124 The mechanisms by which this occurs are not completely understood, but new preclinical studies have shed light on some prevailing theories. Wu et al 125 found that caveolin-1 facilitates T-cell trafficking into the CNS via intercellular adhesion molecule 1 (ICAM-1)-mediated signaling. Caveolin-1 causes acid sphingomyelinase to interact with ICAM-1 increasing the binding affinity for peripheral immune cells. 126 Once activated, ICAM-1 facilitates peripheral immune cell diapedesis into the brain. This process occurs via Src phosphorylation within endothelial cells and a subsequent conformational change to ICAM-1, which directly induces the transcellular migration. 127 The leaky BBB enhances the entire process during sepsis. After entering the brain, T cells are recruited toward damaged glia via cytokine release. Recent evidence suggests that IL-17A aids this migration process. 128 In addition, T cells are helped by astrocytes to re-cross the leaky BBB and carry information about the status of the brain to the rest of the body. 129 It is postulated that the peripheral immune cells also release cytokines that maintain the leakiness of the BBB as they exit the brain. When and how long this cross-talk between microglia and peripheral immune cells persists remains to be elucidated. The feedback loop has, however, been implemented in non-autonomous neuronal death. 130 The brain regions most susceptible are the nigra-striatal pathway and hippocampus. 131 Future studies are warranted to further characterize the brain/immune communication network and, in particular, where the peripheral immune cells ultimately reside after exiting the brain. Collectively, these mechanisms represent the complicated and multifactorial mechanisms that must be involved in sepsis at the BBB. An integrated overview of the brain and peripheral mechanisms found in acute sepsis is shown in Figure 2.

Complex neuroinflammatory processes promote BBB dysfunction in early sepsis which enhance neuronal dysfunction and trigger cognitive impairment. Highly complex and multifactorial mechanisms transduce systemic inflammatory signals to the brain in early sepsis, resulting in neuronal dysfunction and subsequent acute and chronic cognitive impairment. The BBB serves as both a nexus and an interface for these signals. Sepsis begins with systemic infection that evokes an exaggerated host immune response from the recognition of pathogen-associated molecular patterns (PAMPs). 132 In this example, sepsis is triggered by a local lung infection (eg, pneumonia). Systemic proliferation of the infection initiates a hyperinflammatory response that stimulates the production of immune cells, cytokines, and other inflammatory mediators. These inflammatory mediators initiate a cascade of events that either directly or indirectly impact the peripheral microcirculation via cardiovascular autonomic alterations, or activation of the coagulation cascade, and converge on cerebral microvessels and their component BBB endothelial cells. Cerebral hypotension and microthrombus formation also initiate ischemia. The convergence of these events lead to BBB dysfunction, including immune cell infiltration, upregulation of adhesion molecules, increased BBB permeability, activation of cerebral cytokines, cerebral edema, and enhanced neuroinflammation.2,37,88,121 Positive feedback of the disrupted BBB on neuroinflammation and potential amplification of this feedback is indicated by the solid bidirectional arrow (direct feedback) and the dotted arrow (indirect feedback).
Mitochondrial dysfunction
Mitochondrial dysfunction is a common consequence of sepsis. It has been described in a number of studies with substantial evidence pointing toward oxidative stress as a contributing factor. This literature is summarized in several excellent reviews.133–136 The abnormality in the function of the mitochondria plays a role in the development of post-sepsis behavioral, psychological, and cognitive dysfunctions such as SAE.137–139 At the cellular level, reactive nitrogen species (RNS), like NO, and ROS, such as peroxynitrite (ONOO−), inhibit complexes I and IV of the electron transport chain (ETC). This inhibition produces a subsequent decrease in oxygen consumption and permits the buildup of O2− species and the eventual leakage of this species across the ETC along with other ROS/RNS.140–142 The leaked species activate uncoupling proteins that cause an increased H+ (proton) permeability from the inner mitochondria into the mitochondrial matrix to form oxide ions. Ultimately, these ions are converted to water without any adenosine triphosphate (ATP) generation. 143 This process may seem to decrease the number of reactive species from the reaction described above, but the downside of continual proton leakage is an induction of cytopathic hypoxia, a condition whereby mitochondria are unable to use oxygen irrespective of the presence or absence of oxygen. 144 In addition to cytopathic hypoxia, ONOO− causes single-strand DNA breaks at the genomic level; this further exacerbates mitochondrial dysfunction because of the high reliance of oxygen in the reparative process of the damaged DNA. 145 Also, some ROS/RNS enhance both endoplasmic reticulum and mitochondrial membrane permeability, which permits the leakage of calcium and proapoptotic proteins into the cytoplasm.146–148
The course of infection, type and amount of ROS/RNS, and the brain regions where oxidative stress occurs are important when investigating the timing of oxidative damage leading to mitochondrial dysfunction. Recent studies in rats using thiobarbituric acid and protein carbonyls as markers of lipid and protein oxidation, respectively, have suggested that lipid peroxidation is consistent and widely distributed in the hippocampus, cerebellum, and cortex 6 hours post CLP, whereas oxidative damage due to protein oxidation was largely restricted to the hippocampus. Investigators also identified a concomitant imbalance in the antioxidant enzymes superoxide dismutase (SOD) and catalase (CAT). They found that the SOD activity increased in the first 6 hours post sepsis, whereas both SOD and CAT activity levels were decreased compared with sham-injured mice at 12 to 96 hours. 138 Taken together, these findings suggest that oxidative damage in the CNS occurs much earlier than expected in sepsis. Expanding on these findings, Barichello et al used N-acetylcysteine (NAC) and deferoxamine (DFX) antioxidants as a therapeutic intervention in male rats at 6 hours post CLP. They found that the combined administration of NAC and DFX reduced oxidative hippocampal damage, but not when administered separately. 149 These results further emphasize the importance of all CNS antioxidant systems and signify that multiple targets are required for adequate therapeutic efficacy in sepsis.
Overall, the systemic immune response in sepsis accelerates the increased generation of ROS/RNS, which, in turn, promotes lipid peroxidation in the cerebrovasculature and brain parenchyma. The continued assault from the periphery perpetuates a vicious cycle of ROS/RNS generation between the brain and the periphery. As the overproduction of ROS/RNS overwhelms the capacity of the antioxidant system, the end results manifest as neuroinflammation, ischemia, and increased BBB permeability. Most importantly, the vicious cycle promotes an impaired oxidative metabolism which persists throughout the duration of sepsis and likely continues after recovery. 150 Thus, sustained production of ROS/RNS after recovery is hypothesized to be another mechanism that contributes to long-term neurological impairment post sepsis.
Putative role of tissue non-specific alkaline phosphatase at the BBB
The identification of unexplored membrane proteins may be key to better understanding the specific barrier functions of the BBB in disease states such as sepsis. In turn, this knowledge may provide novel therapeutic targets for intervention. One potential therapeutic target localized primarily to the surface of brain endothelial cells is the non-specific isoform of alkaline phosphatase (AP). The enzyme AP has been shown to play an integral role in the regulation of inflammation and can be found either as a soluble form in the peripheral circulation or as a membrane-bound form on brain endothelium, as well as numerous other cell types in the periphery. There are 4 isoforms of AP in humans encoded by 4 separate genes (gene names are in italics): intestinal alkaline phosphatase (IAP; ALPI), placental alkaline phosphatase (PLAP; ALPP), germinal alkaline phosphatase (GCAP; ALPPL2), and tissue non-specific alkaline phosphatase (TNAP; ALP).151,152 TNAP, also known as bone/liver/kidney AP, is the most abundant AP isoform in humans and rodents. TNAP is the only isoenzyme of AP detected in the human brain and has long been used as a marker of brain endothelium, although its presence has also been detected in neurons.153,154
The cellular and molecular mechanisms underlying TNAP’s functional role in brain endothelium and BBB are unclear; however, results from numerous studies across several species strongly suggest that TNAP plays a role in the transport of specific classes of compounds across the BBB. 155 Brain endothelial cell TNAP protein may also help facilitate cross-talk between the BBB and other cell types; in addition, a number of molecules, including cyclic adenosine monophosphate (cAMP) and IL-6, have been shown to modulate TNAP expression. Deracinois et al 153 found that TNAP expression was increased in brain endothelial cells, and that the inhibition of AP activity using levamisole, a non-specific AP inhibitor, increased brain endothelial cell permeability. We speculate that TNAP’s regulatory phosphatase activity on a number of BBB endothelial proteins may play an important role in maintaining BBB integrity, thereby alleviating septic encephalopathy or long-term brain dysfunction. As shown in Figure 3 of our in vivo study, TNAP enzyme activity appears to be upregulated in CLP-injured mice compared with their sham-injured counterparts. However, the mechanistic function of TNAP in the BBB remains to be elucidated in sepsis and is currently being investigated in our lab.
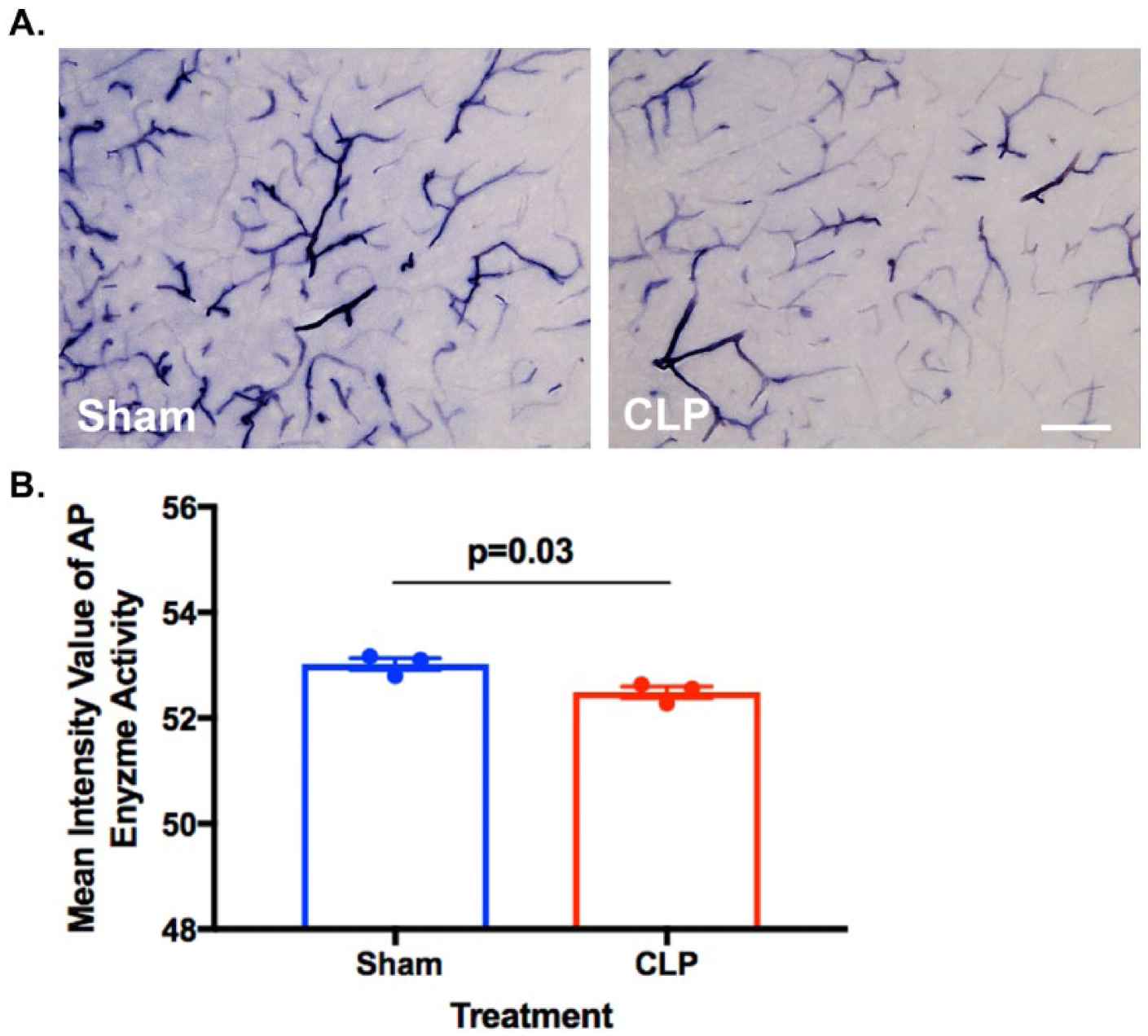
Alkaline phosphatase (AP) activity in the brain and BBB. (A) Histological staining for AP activity shows decreased TNAP enzyme activity in the cortex of septic male mice (10-15 months old) subjected to the cecal ligation and puncture (CLP) model of experimental sepsis. C57BL/6J mice were subjected to CLP or a sham injury and brains were harvested 24 hours later. (B) Graph shows the quantification of cortical AP enzyme activity in CLP (n = 3, 52.49 ± 0.1094) versus sham (n = 3, 53 ± 0.1142) mice (sections = 3 per mouse; data represented as mean ± SEM, *P < .05, t(4) = 3.384, unpaired Student’s t-test, scale bar = 115 µm). AP activity was assessed in 35-µm brain sections with the BCIP/NBT AP Substrate Kit (Vector Laboratories, Burlingame, CA) following previously published methods. 156
Other brain-specific functions of TNAP have been described as having a role in proliferation and migration in the developing nervous system, control of axonal growth formation and maturation of synapse, and dephosphorylation of extracellular phospho-tau in AD.154,157–159 Despite the absence of a clearly elucidated mechanism for TNAP in brain endothelium and neurons, emerging data suggest that the manipulation of the AP activity can influence disease outcomes. For example, pretreatment of experimental autoimmune encephalomyelitis (EAE) mice with bovine intestinal AP reduced the disease severity through a mechanism that caused a reduction in neuroinflammation and autoreactive T regulatory cell proliferation. 160 Increased levels of AP have also been detected in blood of epileptic patients, suggesting its applicability as a potential biomarker for many neurological diseases. 161
Pharmaceutical Interventions in Sepsis
Antimicrobial delivery across the BBB
There is a current shift in the literature regarding specific pathogens that play a role in the inflammatory response associated with sepsis. Knowing the microbe implicated in sepsis best dictates which appropriate antibiotic or other type of treatment is required. Whereas older studies implicate Gram-negative bacteria necessitating antibiotic intervention in sepsis, newer studies have begun to reveal that other pathogens like Gram-positive bacteria, fungi, and viruses can also stimulate the inflammatory response associated with sepsis. According to recent epidemiologic studies, Gram-positive bacteria cause approximately 50 000 more cases of sepsis every year in the United States compared with Gram-negative bacteria.8,14,162 Because factors such as the type of inciting pathogen and the site of infection are good predictors of patient mortality, it is essential that the antimicrobial agent be used to effectively treat the disease as well as any subsequent side effects such as neurological impairment. Currently, this is often difficult and impractical in the hospital, as the time from diagnosis to initiation of treatment is critical. In addition, the sepsis field has faced many difficulties in developing effective therapeutics to treat sepsis. Currently, there are no Food and Drug Administration (FDA)-approved drugs used to treat sepsis as there have been numerous clinical trial failures over the past 15 years.163–166 This difficulty stems from our incomplete knowledge about the mechanisms that underlie the disease pathology associated with sepsis.
Initial suspicion of sepsis necessitates the use of non-specific broad-spectrum antibiotics against Gram-positive (eg, vancomycin) and Gram-negative (eg, imipenem) bacteria before blood cultures become available. Following pathogen identification, the initial antibiotic regimen is often narrowed to a single agent.45,167 Although numerous human and animal studies have shown an increase in survival following antibiotic administration, limited published data are available on whether or how antibiotics are able to penetrate the BBB. More importantly, the effects of antibiotics and other antimicrobials on brain function and sepsis-associated neurological impairment are not well studied.168–173 Thus, a complete knowledge of drug mechanisms in the CNS is essential for identifying an appropriate drug regimen and therapeutic approach to treat the neurological impairment associated with sepsis.174,175
The most common drug classes used to treat sepsis are shown in Table 3. Drugs belonging to the fluoroquinolone and sulfonamide classes, along with rifampin, metronidazole, and chloramphenicol, readily enter the brain regardless of disease state. Antimicrobials that do not normally penetrate the brain may readily cross into the BBB, or into the cerebrospinal fluid (CSF), due to opening of TJs and reduced P-gp activity. 186 In contrast, more hydrophilic and larger drugs such as vancomycin and members of the β-lactam class of antibiotics do not readily enter the CSF or brain unless the meninges are inflamed. 187
Antimicrobials used in sepsis treatment and their corresponding CNS penetration.

CNS, central nervous system.
indicates higher, − indicates lower, and 0 indicates neutral or no change.
Intravenous immunoglobulin administration
Active advancement in understanding the pathophysiology of sepsis has led to the use of emerging immunomodulatory adjuvants to target septic encephalopathy. The acute phase of sepsis is embodied by a diminished production of immunoglobulin G (IgG) because the immune system takes 1 to 2 weeks to generate sufficient IgG levels needed to respond to an infection. 188 Therefore, the observed reduction in IgG levels has warranted the use of immunomodulatory intravenous immunoglobulins (IVIgs) in sepsis patients.188,189 The mechanisms by which IVIg operates are complex and remain unclear.190,191 However, it has been proposed that the IVIg polyclonal IgG domains exert an immunomodulatory function by binding Fc receptors (FcγRs) found on many immune cell types (ie, microglia, endothelial, leukocyte). IVIg binding is thought to neutralize endotoxins/cytokines, inhibit complement activation, and block leukocyte adhesion molecule binding.90,192,193
Several studies have demonstrated the efficacy of IVIg treatment in sepsis. Esen et al 193 showed that the administration of IVIg enriched with IgA and IgM improved BBB permeability, reduced sickness behavior, and improved mortality in CLP-induced rats. Further investigations by the same group revealed that the improvement in BBB integrity, neuronal destruction, and amelioration of septic encephalopathy is mediated by the inhibition of complement 5a (C5a). 90 A small number of clinical meta-analytical studies have reported a decrease in mortality of sepsis patients administered IVIg; this finding complements the results observed by Esen et al. 193,194–196 In contrast, a larger double-blinded randomized control trial conducted in the International Neonatal Immunotherapy Study (INIS) showed no significant differences in mortality following IVIg administration.192,197 Note that the studies included in the meta-analysis showing a decrease in mortality after IVIg administration consisted of relatively small patient populations, which suggests that these studies may not have been sufficiently powered to detect meaningful differences in mortality.191,192,198,199
Taken together, the apparent effects of IVIg in sepsis treatment appear to be promising yet inconclusive. The high cost of treatment combined with unknown mechanism(s) of action and limited efficacy in a number of publications have made it difficult for organizations like the FDA and the Surviving Sepsis Campaign Guidelines (SSCG) to recommend IVIg as an adjuvant in sepsis treatment.188,190,192
Conclusions
The heterogeneous presentation and causes of sepsis are profoundly linked to its variable clinical outcomes. Chronic neurological impairment is an increasingly common yet poorly understood clinical outcome. Understanding the mechanistic determinants of BBB integrity during sepsis is critically important for sepsis diagnosis and implementation of treatment options to ensure a positive prognosis. Importantly, long-term prognosis in sepsis survivors is linked to both transient and permanent alterations in BBB permeability and function. Thus, targeting the BBB should be incorporated as part of a short- and long-term therapeutic strategy in all sepsis patients. The development of therapies that inhibit BBB dysfunction and stimulate normal BBB function will limit mortality, suppress neuroinflammation, and improve neurological outcomes in sepsis survivors. Equally important for effective sepsis treatment is a better understanding of how antimicrobials and other drugs (eg, IVIgs or vasopressors) used in treating sepsis readily cross the BBB, and whether there are any additional unknown impacts on brain function. Taken together, the identification of cellular and molecular mechanisms that preserve BBB function in the face of sepsis will provide valuable therapeutic targets to treat numerous inflammatory disorders that target both the brain and the periphery—ranging from AD and stroke to diabetes and cardiovascular disease.
Footnotes
Funding:
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
DCN and CMB contributed to manuscript design, compilation of manuscript, and creation of figures and tables. ALB wrote section on tissue non-specific alkaline phosphatase (TNAP). ASM contributed to the table on antimicrobials used in sepsis. JG contributed to the section on BBB overview. BPL-W contributed to the section on neuroinflammation and BBB permeability. SAB performed tissue alkaline phosphatase (TNAP) enzyme stain and image analysis. WJG contributed to the section on neuroinflammation and BBB permeability. PRL contributed to section on pharmacological intervention in sepsis. All authors reviewed the final documents and provided comments.