
Abstract
Multisystem Inflammatory Syndrome in Children (MIS-C), representing a new entity in the spectrum of manifestations of COVID-19, bears symptomatic resemblance with Kawasaki Disease (KD). This review explores the possible associations between KD and the human coronaviruses and discusses the pathophysiological similarities between KD and MIS-C and proposes implications for the pathogenesis of MIS-C in COVID-19. Since 2005, when a case-control study demonstrated the association of a strain of human coronavirus with KD, several studies have provided evidence regarding the association of different strains of the human coronaviruses with KD. Thus, the emergence of the KD-like disease MIS-C in COVID-19 may not be an unprecedented phenomenon. KD and MIS-C share a range of similarities in pathophysiology and possibly even genetics. Both share features of a cytokine storm, leading to a systemic inflammatory response and oxidative stress that may cause vasculitis and precipitate multi-organ failure. Moreover, antibody-dependent enhancement, a phenomenon demonstrated in previous coronaviruses, and the possible superantigenic behavior of SARS-CoV-2, possibly may also contribute toward the pathogenesis of MIS-C. Lastly, there is some evidence of complement-mediated microvascular injury in COVID-19, as well as of endotheliitis. Genetics may also represent a possible link between MIS-C and KD, with variations in FcγRII and IL-6 genes potentially increasing susceptibility to both conditions. Early detection and treatment are essential for the management of MIS-C in COVID-19. By highlighting the potential pathophysiological mechanisms that contribute to MIS-C, our review holds important implications for diagnostics, management, and further research of this rare manifestation of COVID-19.
Background
On May 14, 2020, the CDC issued an advisory regarding a disease with similar symptoms to Kawasaki Disease (KD) in pediatric patients exposed to SARS-CoV-2 (severe acute respiratory syndrome coronavirus 2). However, despite being life-threatening, the disease is considered a rare complication of SARS-CoV-2 infection. 1 The WHO classified this Kawasaki-like disease as “multisystem inflammatory syndrome in children and adolescents temporally related to COVID-19 (MIS-C)” (also known as Pediatric Multisystem Inflammatory Syndrome temporally associated with SARS-CoV-2; PIMS-TS). 2 With the COVID-19 (coronavirus disease 2019) pandemic looming large, the emergence of MIS-C has brought renewed attention and interest to KD. 1
While the specifics are yet unknown, the pathogenesis of KD is commonly thought to involve a post-infectious etiology. 3 It has been repeatedly hypothesized that there exists a connection between respiratory viruses and Kawasaki Disease, 4 with positive viral polymerase chain reaction (PCR) results frequently seen in KD patients. 5 Moreover, some studies propose the existence of a hitherto unidentified respiratory virus (the “KD agent”).4,6 The viral theory of KD pathogenesis is supported by the epidemiological, clinical, histopathological, and laboratory features of KD. 3 One group of respiratory viruses mentioned in association with Kawasaki-like symptoms are the human coronaviruses (HCoVs). 7 Moreover, though KD and MIS-C are separate entities that differ when it comes to age and geographic areas, similarities exist in how they present clinically, as is summarized in Table 1. This review summarizes the literature exploring the association and commonalities between KD and the human coronaviruses and discusses the implications for the pathogenesis of MIS-C in COVID-19.
Similarities and differences in MIS-C and KD.

Abbreviations: KD, Kawasaki disease; LV, left ventricle; MIS-C, multisystem inflammatory syndrome related to COVID-19.
The Human Coronaviruses and KD
In 2005, Esper et al 7 reported a possible association of a strain of HCoV-NL63 (termed New Haven Coronavirus) with KD in a case-control study of 11 KD patients and 22 controls (72.7% of KD patients NL63+ vs 4.5% of controls NL63+; P = .015).Subsequently, though several case-control studies have explored the association of coronaviruses with KD, there has been a great deal of conflicting evidence4,5,7,11-16 (Table 2). The differences in results may be due to differences in geographical location, study population, study design, sampling (timing and method of collection, handling, and testing of samples), criteria for selection of cases (inclusion of exclusion of patients with incomplete/atypical KD) and control patients, and coronavirus species tested for.4,5,7,11-22 Furthermore, case series by Shimizu et al, 17 Baker et al 21 (both USA and The Netherlands), Chang et al 18 (Taiwan) and Cho et al 20 (South Korea) have reported HCoV positivity rates ranging from 0% to 2% in KD patients. A case report by Bohlok et al 19 (Lebanon) in 2020 also described atypical KD following HCoV-OC43 infection in 13.5-year-old male, while one by Giray et al 22 (Turkey) in 2016 described HCoV-OC43/HKU1 infection in a 17-month-old female. Interestingly, Rowley et al 23 identified a “corona” of “spikes” surrounding viral particles in bronchial epithelium visualized using transmission electron microscopy, a description that is reminiscent of the crown-like projection of spike proteins that gives the coronaviruses their name. 23 However, despite more than a decade of research exploring the association of the HCoVs with KD, no conclusive evidence has been discovered.
Case control studies exploring the association of Human Coronaviruses with KD.
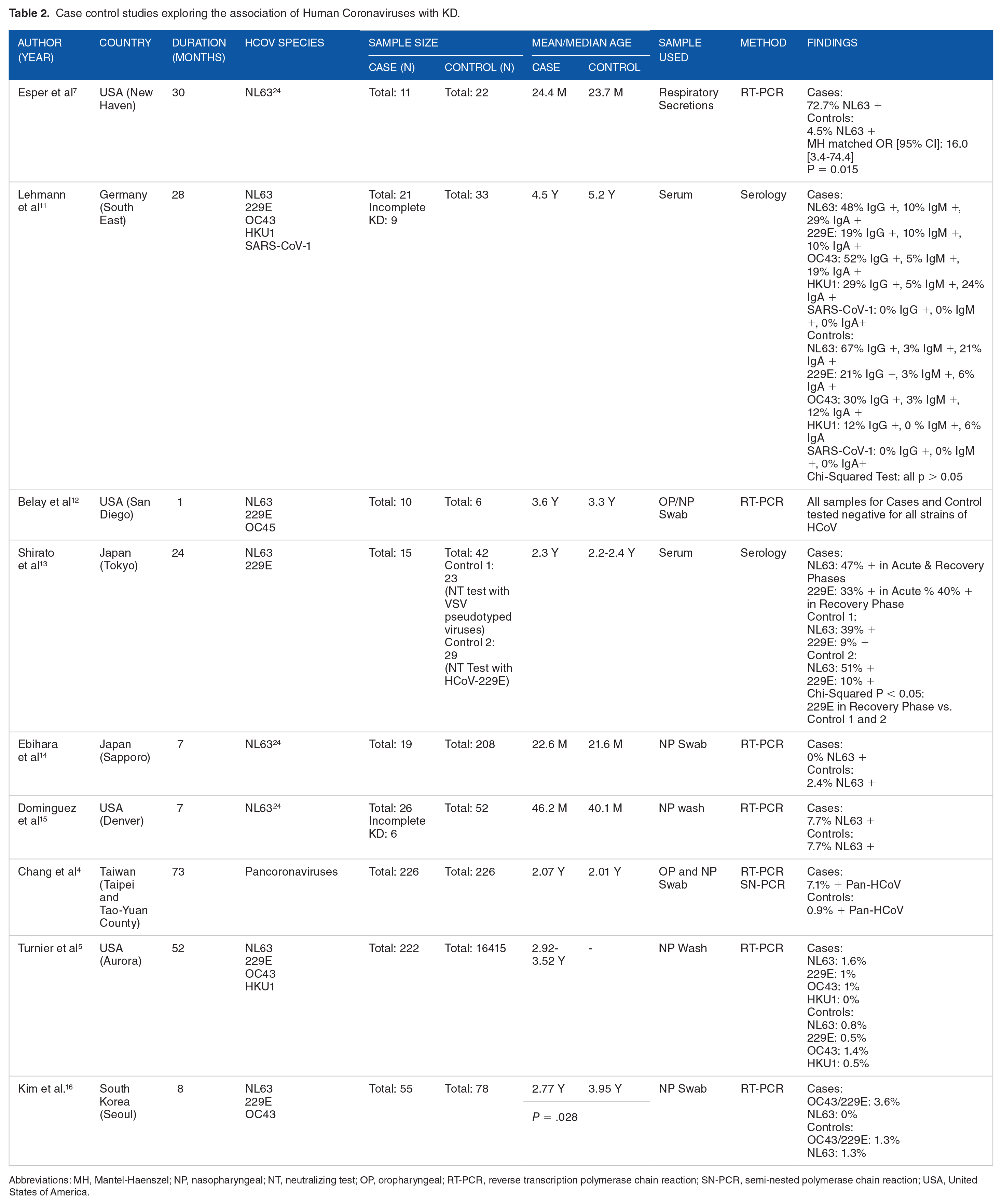
Abbreviations: MH, Mantel-Haenszel; NP, nasopharyngeal; NT, neutralizing test; OP, oropharyngeal; RT-PCR, reverse transcription polymerase chain reaction; SN-PCR, semi-nested polymerase chain reaction; USA, United States of America.
COVID-19’s Cytokine Storm
Cytokines is a broad term to describe a category of proteins that play a key role in cell signaling and the immune system. Homeostasis is maintained by the body by a balance between pro- and anti-inflammatory cytokines. In KD, it is proposed that abnormal activation of the immune system results in the release of pro-inflammatory cytokines. 25 Similarly, COVID-19 has been shown to cause a massive release of pro-inflammatory cytokines (“cytokine storm”) and immune dysregulation, increasing disease severity24,26 and possibly precipitating multi-organ failure. 27 Pro-inflammatory cytokines activate more immune cells and promote leukocyte extravasation, causing local tissue damage and vasculitis. 25 Amongst these cytokines is believed to be TNF-α, which mediates elastin breakdown and eventual aneurysm formation. 28 IL-6 is thought to play a major role, as is supported by its increased levels in MIS-C 29 and the effectiveness of tocilizumab (IL-6R antagonist) in treating it. 30 IL-6 expression is increased by TNF- α, 30 and its effects include promoting CD8+, Th17, and self-reactive CD4+ cells, while inhibiting Treg cells. 31 NF-κB in COVID-19’s cytokine storm induces the IL-6 amplifier (IL-6 Amp), which results in a positive feedback loop of further pro-inflammatory cytokine release, 32 and also induces TNF- α. 32 Moreover, a study demonstrated increased proinflammatory Th17 cell activity with concomitantly decreased anti-inflammatory Treg cell function in KD. 33 This change in Th17 and Treg activity is akin to what is seen in KD, 33 and murine models have shown that inhibition of Th17 cytokines via JAK2 inhibitor Fedratinib may prevent the adverse outcomes of Th17-associated cytokine storm in COVID-19. 34 Thus, understandably, most MIS-C cases have demonstrated recovery after treatment with immunomodulatory agents, including anti-TNF and IL-6 inhibitors. 35
Superantigenic Behavior of SARS-CoV-2
There has been long-standing speculation that the pathogenesis of KD might involve superantigens that cause immune dysregulation, and ultimately, a cytokine storm. 36 Though SAGs are classically bacterial, known and potential viral SAGs include in mouse mammary tumor virus (MMTV), CMV, and EBV. 36 Cheng et al 37 used structural-based computational models to demonstrate the possible superantigenic activity of SARS-CoV-2,and noted that a rare mutation due to a single amino acid substitution from a European strain of the virus resulted in stabilization and stronger binding of SARS-CoV-2’s spike protein with TCR Vβ. 37 Considering this, it is possible that the superantigenic activity of SARS-CoV-2 could contribute to the cytokine storm that characterizes the MIS-C. 38 Table 3 summarizes the cytokine and superantigenic associations in KD and COVID-19.
Cytokines and superantigens in COVID-19 and Kawasaki disease.
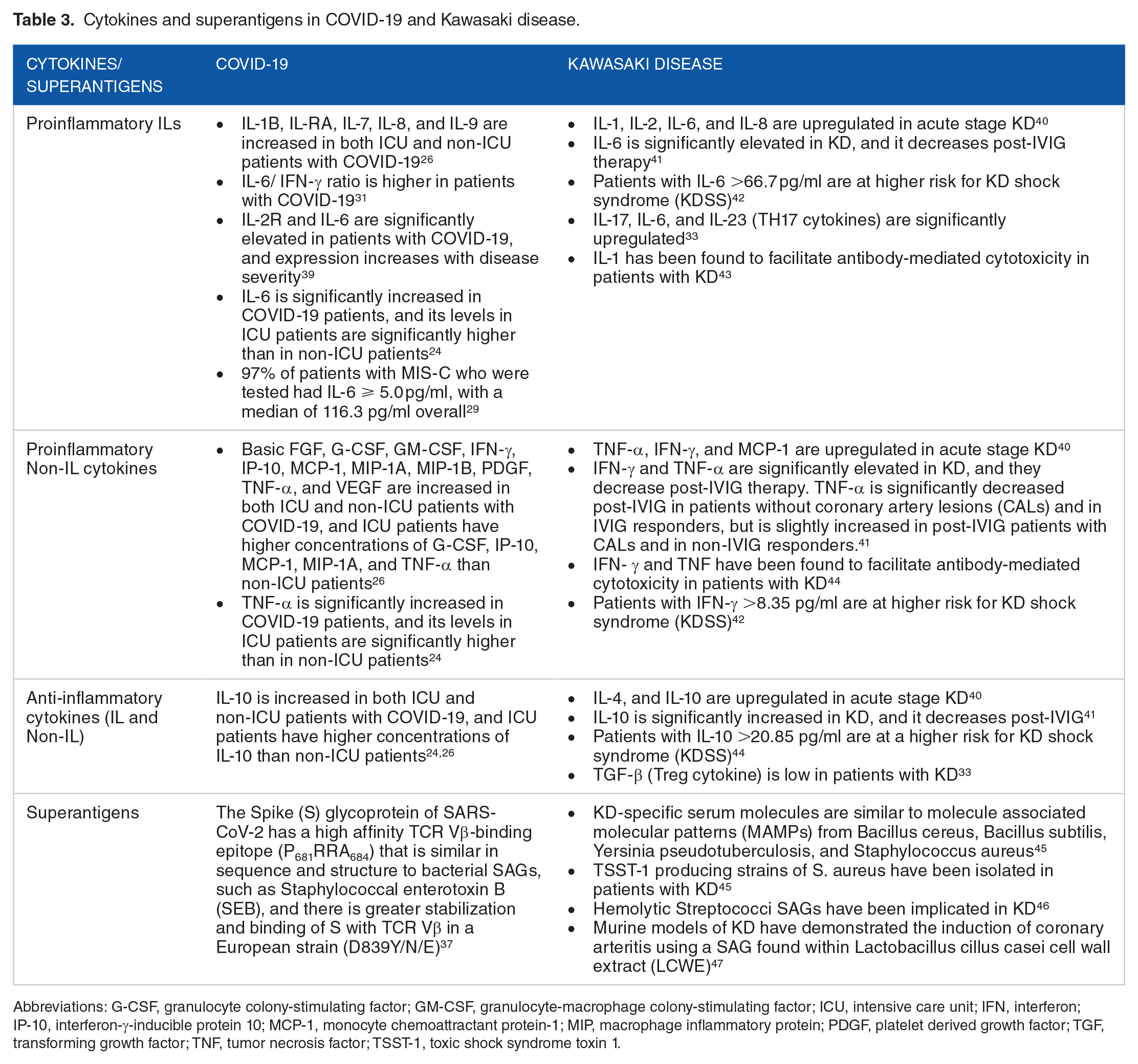
Abbreviations: G-CSF, granulocyte colony-stimulating factor; GM-CSF, granulocyte-macrophage colony-stimulating factor; ICU, intensive care unit; IFN, interferon; IP-10, interferon-γ-inducible protein 10; MCP-1, monocyte chemoattractant protein-1; MIP, macrophage inflammatory protein; PDGF, platelet derived growth factor; TGF, transforming growth factor; TNF, tumor necrosis factor; TSST-1, toxic shock syndrome toxin 1.
Genetics: Possible Connections Between SARS-CoV-2 and KD
Genetics are widely considered to play a role in the susceptibility to KD. 48 Genome-wide association studies have demonstrated several gene variants associated with KD. These include FCGR2A (FcγRII), ITPKC (inositol 1,4,5 triphosphate kinase C), CD40, CASP3 (Caspase 3), multiple alleles of HLA I and II (Human Leukocyte Antigens I and II), CD209 (CD209/DC-SIGN), and BLK (B lymphoid tyrosine kinase). 48
Due to the nature of the novel coronavirus, genetic studies regarding SARS-CoV-2 are still nascent. However, a few potential developments have been made. HLA associations have been demonstrated, with HLA-B*46:01 having the lowest number of predicted binding peptides for SARS-CoV-2. 49 This indicates that individuals with the allele may be particularly vulnerable to COVID-19, as was the case previously with SARS-CoV-1. 49 Moreover, HLA-B*15:03 demonstrated the greatest ability for presenting highly conserved SARS-CoV-2 peptides shared among the HCoVs, suggesting a possibility that it may provide cross-protective T-cell-based immunity. 49 Although neither HLA-B*46:01 nor HLA-B*15:03 have been associated with KD, HLA-B*54:01 has been found to increase susceptibility to the disease. 50 A small proportion of COVID-19 patients develop features of a cytokine release syndrome (CRS) 1-2 weeks after onset of infection. Recent evidence suggests that IL-6 may be a key mediatory in the CRS associated severe SARS-CoV-2 infection, 51 and it is hypothesized that IL-6 polymorphisms may predispose to severe COVID-19. 52 Increased IL-6 levels also play a role in the acute hyperinflammatory response in KD, showing a potential commonality between SARS-CoV-2 infection and KD. 51
Since SARS-CoV-2 bears considerable genetic similarity to SARS-CoV-1, it is in the realm of thought that genetic associations with SARS-CoV-1 may hold true to some extent for SARS-CoV-2 as well. Assuming the association holds true, we can postulate genetic links between SARS-CoV-2 and KD. To begin with, there is an association between the FcγRIIA-R/R131 genotype and a severe course of SARS-CoV-1, 53 and the FCGR2A (FcγRII) has been found to be associated with increased susceptibility to KD. 48 Similarly, a single nucleotide polymorphism in the CD209 promoter is associated with severe SARS-CoV-1 infection, 54 while the CD209 gene has been shown to be associated with increased susceptibility to KD. 48 The +874 A/T polymorphism in the IFN-γ (interferon gamma) is associated with a higher risk of developing SARS-CoV-1 infection, 55 and the IFN-γ gene has been identified in a mouse model as a candidate gene for association with KD through its regulatory cytokine-suppressing inflammatory response. 28
Currently, there is insufficient genetic research pertaining to SARS-CoV-2 to support any associations with KD, and the links remain highly speculative. However, based on previous experience with SARS-CoV-1 and MERS, MBL (Mannose-Binding Lectin), CD147, CCL2, IL-12, and HLA genes have been highlighted for consideration in playing a role in susceptibility to SARS-CoV-2 and severe COVID-19. 56 With the initiation of the COVID-19 Host Genetics Initiative, 57 the interplay of genetics with SARS-CoV-2 infection and COVID-19 will be increasingly unraveled. This will serve to shed more light upon the genetic associations between KD and SARS-CoV-2.
Antibody-Dependent Enhancement
Antibody-dependent enhancement (ADE), the phenomenon whereby dysfunctional antibodies amplify viral entry and infection instead of diminishing it, has been demonstrated in the past with viruses, such as HIV 58 and the coronaviruses, including SARS-CoV-1 and MERS-CoV.59,60 The most common mechanism of ADE is Fc receptor (FcR)-dependent, where binding of FcR to the virus-antibody complex augments the attachment between the cell and the virus, thus resulting in enhanced infectivity. 58 ADE may also occur via binding of virus-antibody complexes to complement receptors or by induction of a conformational change in the viral glycoproteins, resulting in enhanced fusion. 58
SARS-CoV-1 has demonstrated ADE in several in vitro experiments. Antibodies generated in response to a SARS-CoV-1 in a hamster model enhanced FcγRII (CD32)-dependent viral entry into B cells. 59 Another study using anti-Spike immune serum reinforced that ADE in SARS-CoV-1 is predominantly dependent on FcγRII (CD32). 61 This study also showed that certain immune cells are only infected by SARS-CoV-1 in the presence of anti-Spike immune serum. 61 A different study used an HL-CZ human pneumocyte cell line isolated from a leukemia patient to demonstrate ADE. 62 The HL-CZ cells expressed both ACE2 as well as higher levels of FcγRII (CD32), and when they were treated with diluted anti-sera collected from SARS-CoV-1 patients, there was increased viral infectivity, as opposed to when they were treated with a more concentrated anti-sera. 62 Furthermore, a study found that anti-Spike antibodies increased infection in macrophages, and this depended on an intact FcγRII extracellular and intracellular signaling domain. 63 FcγRII with mutated intracellular and normal extracellular domains were unable to trigger ADE, despite being able to bind to the ligand. 63
Similarly, MERS-CoV has also shown ADE in vitro. Wan et al. 60 demonstrated a novel mechanism for ADE in MERS-CoV using Mersmab 1, a monoclonal antibody that binds to the receptor-binding domain of MERS-CoV’s Spike-ectodomain (S-e). Mersmab 1 was shown to bind MERS-CoV S-e with the Fc receptors on human HEK293T (human kidney) cells. Controlled experiments demonstrated that Mersmab1 mediated viral entry into HEK293T cells with exogenously expressed Fc-receptors as well as into macrophages with endogenously expressed Fc-receptors, but blocked viral entry into cells expressing MERS-CoV’s receptor dipeptidyl peptidase-4 (DPP-4). The ADE varied with the dosage of Mersmab 1, and with the expression of viral receptors and Fc-receptors. Cells with both Fc-receptors and DPP-4 had the highest viral entry at the intermediate dose. Moreover, the study demonstrated that neutralizing antibodies mimic the function of viral receptors, binding to the spike protein and triggering a conformational change in it, which results in sequential proteolytic cleavage, thus mediating viral entry into cells expressing FcR.
Various studies in the past have found that there is significant cross-reactivity between antibodies generated in response to different coronavirus strains, suggesting the possibility of non-specific ADE. One such study found that 7/28 (25%) of SARS-CoV-1 patients had cross-reactive neutralizing anti-MERS antibodies, 64 while another found appreciable cross-reactivity between MERS-CoV with 3 out of 4 of the seasonal human coronaviruses (HKU1, 229E, OC43, and NL63 CoV). 65 Moreover, serum samples from COVID-19 patients cross-react with nucleocapsid antigens of SARS-CoV-1, 66 while antibodies against the Spike protein and receptor-binding domain (RBD) cross react with plasma from SARS-CoV-1 patients. 67 The cross-reactivity is mostly with the non-RBD regions, resulting in non-neutralizing antibodies. 67
As MIS-C occurs late after infection with SARS-CoV-2, once antibodies have developed, it may be possible that ADE may contribute to the underlying mechanism of a pro-inflammatory cytokine storm, yet this remains to be explored.35,68 This speculation is supported by the geographical distribution of MIS-C, as prior outbreaks in areas with different coronavirus strains may prime for ADE due to antigenic epitope heterogeneity. 68 In the in vitro model that demonstrated ADE of SARS-CoV-1 in HL-CZ cells, the cells were found to have increased pro-inflammatory cytokine secretion. 62 A study using SARS-CoV-1/macaque models 69 demonstrated that anti-spike IgG in infected lungs prior to viral clearance resulted in recruitment of proinflammatory macrophages and a 5–10 fold increase in IL-6, MCP1, and IL-8. 69 Lastly, since by adulthood most individuals develop natural immunity against coronaviruses, the resultant antibodies may be passed to the infant transplacentally and via breastfeeding, and may cross-react with SARS-CoV-2. With age and with weaning, as the titers of these passive antibodies fall, ADE may occur in the presence of SARS-CoV-2 infection. Studies have highlighted this dose-dependent correlation between antibodies resulting in ADE.60,62 Such a phenomenon has been observed with other viruses such as the Dengue virus, in which infants <1 year of age can develop dengue hemorrhagic fever with a primary dengue virus infection due to the maternal dengue-specific antibodies. 70
Complement activation
Due to its ability to cause inflammation, the complement pathway may be involved in the pathogenesis of MIS-C in COVID-19. A study found that the nucleocapsid (N) proteins of SARS-CoV-1, MERS-CoV, and SARS-CoV-2 bind to MASP-2, the serine protease in the lectin pathway of the complement cascade, causing inflammatory lung injury. 71 The study also found complement hyper-activation in COVID-19 patients, and observed a promising suppressive effect when the patients were treated with anti-C5a monoclonal antibody. 71 Moreover, in the advanced stages of COVID-19, C3 inhibition has the potential to broadly control systemic inflammation affecting the microvascular beds of vital organs of the body. 72 A study exploring complement-associated microvascular injury in cases of severe COVID-19 found striking septal capillary injury, in addition to extensive deposits of terminal complement complexes (C5b-9) in the lungs. 73 As the defining feature of KD is vasculitis and general systemic inflammation, further investigation into the role of complement in its pathogenesis is warranted.
Endotheliitis
Being a multisystem inflammatory disease that affects mainly the medium-sized arteries, KD is known to cause endotheliitis. 74 Angiotensin Converting Enzyme 2 (ACE2) receptors are present in arterial cells of several organs such as the lungs, heart, kidneys, and intestines.75-77 As ACE2 receptors promote SARS-CoV-2′s entry into cells and its replication, 75 SARS-CoV-2 may cause endotheliitis via endothelial ACE2 receptors. 76 Viral inclusion bodies in capillary endothelial cells, along with cellular infiltrates and apoptosis of endothelial cells, confirm endotheliitis in SARS-CoV-2 infection. 76 The endotheliitis and resultant endothelial dysfunction may lead to tissue edema, a procoagulant state, and potentially, multi-organ dysfunction. 77 Thus, SARS-CoV-2 infection and inflammation in COVID-19 may function as “priming triggers” that accelerate KD development, and highlight an important possible connection between COVID-19 and KD. 74 Given this, it can be hypothesized that endotheliitis may contribute to the pathophysiological mechanism of association between KD and MIS-C.
Reactive oxygen species
Given the multisystemic inflammatory nature of KD, it has been suggested that enhanced oxidative stress may contribute to the pathogenesis of the disease. 78 The overproduction of ROS upregulates cytokine/chemokine gene expression, raising serum levels of TNF-α, IL-1, and IL-6. 79 In addition, increased ROS may lead to activation of kinase pathways and redox-sensitive transcription factors, such as NF-κB, which further upregulate cytokines. 79 These amplify the inflammatory process and cause tissue migration of neutrophils and macrophages. 79 In KD, the increased oxidative stress and resultant cytokine release may promote the vasculitic changes and characteristic coronary artery abnormalities. 79
Increased ROS production may play a role in the development of MIS-C. Viral infections of the respiratory system are often associated with pathophysiological processes culminating in oxidative stress through dysregulation of antioxidant mechanisms. 80 In SARS-CoV-2 infection, inflammatory changes may lead to a dysregulated anti-oxidant system, causing overproduction of reactive oxygen species (ROS), which could contribute to the pathogenesis of COVID-19 and its progression into severe disease.80,81 In addition, inflammation associated with SARS-CoV-2 may also cause macrophages and neutrophils to increase their production of ROS. 82 The levels of angiotensin II (ATII) have been shown to increase linearly in SARS-CoV-2 infection, 83 and this increase mediates an NADH/NADPH oxidase-induced production of ROS. 81 Regardless of the source, increased levels of ROS may ultimately cause significant damage to the vascular endothelium in COVID-19. 81
Conclusion
MIS-C represents a new entity in the spectrum of manifestations of COVID-19, with cases being reported from all over the world. It is important to understand the pathophysiological mechanisms behind this new condition, as this will guide progress toward diagnostics and treatments. Building on the clinical similarities between KD and MIS-C, our review aimed to compare pathophysiological mechanisms between the2 conditions in order to shed light on this rare manifestation of COVID-19.
Given the history of association of KD with coronaviruses and other viruses,4,5,7,11-22 this review demonstrates how the emergence of MIS-C may not be a wholly unique and unprecedented phenomenon. KD and MIS-C share a range of commonalities, including in their clinical presentation and proposed pathophysiology. Though there is no definitive genetic link between the two, the possibility of one cannot be excluded. Both conditions share features of a hyper-inflammatory syndrome or cytokine storm,25,27 leading to a downstream inflammatory response, and to systemic oxidative stress, that may cause vasculitis and precipitate multi-organ failure.77,80,81 It is possible that antibody-dependent enhancement, a phenomenon seen in previous coronaviruses, may play a role in advancing the cytokine storm in COVID-19.59-63 Additionally, our review also highlights the possible role of the superantigenic behavior of SARS-CoV-2, and of the complement cascade in contributing toward the multi-system inflammatory features of MIS-C. Lastly, given the rarity of MIS-C, this review also speculates, based on evidence, that geographic and genetic factors may predispose to this uncommon manifestation of COVID-19 (Figure 1). The potential immunological and molecular mechanisms for the pathophysiology of MIS-C, as discussed in this review, are summarized in Figure 2. The clinical similarities between MIS-C and KD, as well as the potential underlying pathophysiology in both conditions has important implications when it comes to treatment options, which are contrasted in Table 4.

Proposed model for pathogenesis of MIS-C in COVID-19.

Immune responses and viral mechanisms in the pathogenesis of MIS-C.
Summary of established and potential management options for MIS-C and KD.

Abbreviations: IVIG, intravenous immunoglobulins; IL, interleukin; KD, Kawasaki disease; LMWH, low molecular weight heparin; MIS-C, multisystem inflammatory syndrome related to COVID-19; TNF, tumor necrosis factor.
Early detection and treatment are essential for the management of MIS-C in COVID-19. The WHO has published a preliminary case framework for diagnosis, and has suggested physicians suspect this condition when presented with a patient who displays some or all features of KD or of toxic shock syndrome. 2 Treatment options include immunomodulatory agents, such as IV immunoglobulins (IVIG) and corticosteroids, which have shown positive results. 35 As the pandemic continues to challenge healthcare systems with unprecedented disease manifestations, it is vital that the medical community be prepared to meet this challenge with evidence-based management responses. By highlighting the potential pathophysiological mechanisms that contribute to the condition, our review holds important implications for the diagnosis and management of the MIS-C.
Footnotes
Acknowledgements
We also acknowledge the Research and Development Wing of The Society for Promoting Innovation in Education (SPIE) for providing mentorship to author Ronika Devi Ukrani (medical student) on this project. SPIE is actively involved with innovation, education and research in the academic and public health sector.
Funding:
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Declaration of Conflicting Interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ Note
M Rizwan Sohail is now affiliated to Section of Infectious Diseases, Baylor College of Medicine, Houston, TX, USA.
Author Contributions
FFS and RSM conceived the idea for this review. AU, RDU, FFS and RSM performed the literature search and data extraction. The article was written by FFS, RSM, AU and RDU, and critically reviewed by KJ, MRS and EK. FFS and RSM produced the figures in this article. The final manuscript was approved by all authors.
Ethical Standards
This review was exempt from ethical approval by the institutional review board at the Aga Khan University Hospital, Pakistan.