
Abstract
Neonatal cholestasis (NC) is a diagnostic dilemma frequently countered in a neonatal care unit. Early diagnosis is vital for achieving an optimal patient outcome as many causes of cholestasis such as biliary atresia are time-sensitive and amenable to treatment if analyzed and treated early. Nonetheless, it is not generally simple to analyze these cases right on time as some of them are regularly missed due to the presence of pigmented stools, lack of newborn metabolic screening, and named as instances of prolonged jaundice. In this manner, we prescribe to explore all reasons for prolonged jaundice stretching out past 14 days in neonates. Besides, we suggest that stool card ought to be a piece of release rundown for all newborn children being released from the nursery. This is of most extreme significance in the nation like India where guaranteeing customary follow-up is as yet a tough assignment. These stool cards will help in the early determination of patients with NC particularly biliary atresia and guarantee their auspicious cure. Another reason which needs exceptional say is parenteral nutrition–associated liver illness, as the proportion of preterm babies is getting greater and greater with better neonatal care. These extreme preterm infants are in the requirement for prolonged (>14 days) total parenteral nourishment because of which they are at high hazard for NC contrasted with their more developed peers. In this survey, we will give an understanding of clinical approach, differential diagnosis, and clinical review of NC.
Keywords
Definition and Epidemiology
The term cholestasis refers to diminished bile formation and/or excretion, and the term “neonatal cholestasis” is often used to refer to conjugated hyperbilirubinemia that is present either at birth or develops within the first few months of life. Conjugated hyperbilirubinemia is classically defined as conjugated bilirubin greater than 20% of total serum bilirubin with a minimum level of 1 mg/dL. Bile is formed in the liver and is a blend of bile acids, bilirubin, and fats. It is secreted into canaliculus; from that point, it flows into bile ducts and is at last discharged into the intestine after transient stockpiling inside the gallbladder. Alteration in the normal stream of bile brings about the unusual aggregation of bile salts, bilirubin, and lipids in liver and the blood. Despite the fact that direct hyperbilirubinemia and cholestasis are 2 distinct terms, the simple accessibility, minimal effort, low cost, and abnormal retention of conjugated bilirubin in neonatal cholestasis (NC) make it a reliable surrogate marker of cholestasis.1–3 The overall incidence of NC is reported to be around 1 in 2500 live births. In India, NC constitutes approximately 30% of hepatobiliary disorders in India. By and large, a baby of NC presents at 4.5 weeks of age to a doctor for seeking therapeutic care in India and the average age of presentation to a tertiary center like ours in India is 3.5 months (range: birth to 15 months). Even in developed countries such as Germany and United States, the average age at diagnosis is 60 days. Furthermore, lack of adequate diagnostic facilities and expertise in diagnosing and managing such cases adds to the delay and leads to compromised patient care. All these factors taken together culminate in higher preventable morbidity and mortality related to NC. Even though high-performance liquid chromatography is viewed as the highest quality level, many centers across the world still use the diazo method for evaluating bilirubin, which tends to overestimate the direct fraction at lower bilirubin levels.4–7
Clinical Profile
Presentation of NC is protean, extending across yellowish discoloration of skin to acute liver failure and death. A thorough head-to-toe examination is a must for any case of NC. Acholic stools and high-colored urine are the characteristic terms used to describe an infant with cholestasis (Figure 1). Stools should always be pressed into a paste to obtain its true color. However, these features may not generally be available, and an infant may likewise present with bleeding diathesis; pruritis; deficiency of vitamins A, D, E, K; and failure to thrive. Besides these general symptoms, there are specific clinical features depending on the cause. Coagulopathy may be caused by vitamin K deficiency, liver failure, or severe metabolic derangement of the liver (as in neonatal hemochromatosis). Splenomegaly can be seen in infants who have cirrhosis and portal hypertension, intrauterine infections, storage diseases, and hemolytic disorders such as Rh-isoimmunization. Neurologic abnormalities including irritability, lethargy, poor feeding, hypotonia, or seizures can indicate sepsis, intracranial hemorrhage, metabolic (including Zellweger syndrome) and mitochondrial disorders, or severe liver dysfunction resulting in hyperammonemia and encephalopathy. Low birth weight, thrombocytopenia, petechiae and purpura, and chorioretinitis are regularly associated with intrauterine infections. Congenital infections may also be associated with rash, microcephaly, intrauterine growth restriction, and intracranial calcification. Facial dysmorphism may suggest a chromosomal abnormality or Alagille syndrome. An obvious mass in the upper quadrant of the abdomen may indicate a choledochal cyst. A cardiac murmur increases the likelihood of Alagille syndrome or biliary atresia. On physical examination, infants with biliary atresia are generally thriving well and are appearing well except for jaundice, and stools are often acholic. However, biliary atresia may present with features of advanced liver disease such as ascites and hepatosplenomegaly if there is a delay in diagnosis. By contrast, infants with infectious and metabolic cause appear sick and have inadequate weight gain. 8 A peculiar odor of body or urine may point to a metabolic cause. Examination of male genitalia and ability to fix and follow a moving object may be useful clues for panhypopituitarism and septo-optic dysplasia, respectively.

Acholic stools due to inspissated bile syndrome in a 2-month-old neonate with Rh hemolytic disease.
Etiology
A myriad of potential causes have been identified causing NC and this list is ever increasing with improvement and upgradation of diagnostics and laboratory services. Causes of NC can be broadly divided into surgical and medical causes. Overall biliary atresia is the most common identifiable cause of NC.7–14 According to a latest published meta-analysis comprising 17 studies encompassing 1692 infants, idiopathic neonatal hepatitis was observed in 26%, extrahepatic biliary atresia in 26%, infection in 12%, TPN-associated cholestasis in 6.5%, metabolic disease in 4.37%, α1-antitrypsin deficiency in 4.14%, and perinatal hypoxia/ischemia in 4% of infants with NC. Among the infections and metabolic disorders, cytomegalovirus (CMV) and galactosemia (37% of metabolic disorders) were the most common identifiable causes, respectively. 11 Lack of uniform protocol for diagnosing NC was the main drawback of study (Table 1).
Causes of neonatal cholestasis.

Source: Copyright: Dr Aakash Pandita.
Diagnostic Approach
Joint recommendation by the North American Society for Pediatric Gastroenterology,3,8,15–35 Hepatology, and Nutrition and the European Society for Pediatric Gastroenterology, Hepatology, and Nutrition 2016, has defined abnormal direct/conjugated bilirubin as a serum value >1.0 mg/dL (17 mmol/L), because it is physiologically and clinically complex to consider incorporating consideration of whether or not the direct fraction exceeds 20% of the total bilirubin level as mentioned in some publications.
14
In cases of preterm and small for gestation, with a negative septic and metabolic screen and a normal ultrasound abdomen, a close observation period till the corrected age of 40 weeks or weight of 2.5 kg can be considered, respectively. Commonly employed investigations in a patient of NC are given in Table 2. The aim of the evaluation is to look for treatable causes at the earliest and start specific treatment as appropriate depending on history and clinical evaluation. In general, hepatocellular etiology or hepatitis is associated with a rise in liver enzymes and extrahepatic causes are associated with rise in γ-glutamyl transpeptidase with few exceptions. To access the hepatocellular damage, one needs to do a serum albumin and coagulation profile (prothrombin time, international normalized ratio). Common treatable causes of NC include sepsis (bacterial; viral; CMV, HSV; fungal; etc), thyroid disorder, galactosemia, and hemochromatosis.
Diagnostic workup for neonatal cholestasis.

Source: Copyright: Dr Aakash Pandita.
We provide a schematic diagram to work up a patient of NC syndrome in Figure 2. All patients with age more than 60 days who have previously been admitted and have still not being diagnosed should be referred to a tertiary center with facility for liver transplant for optimal management.
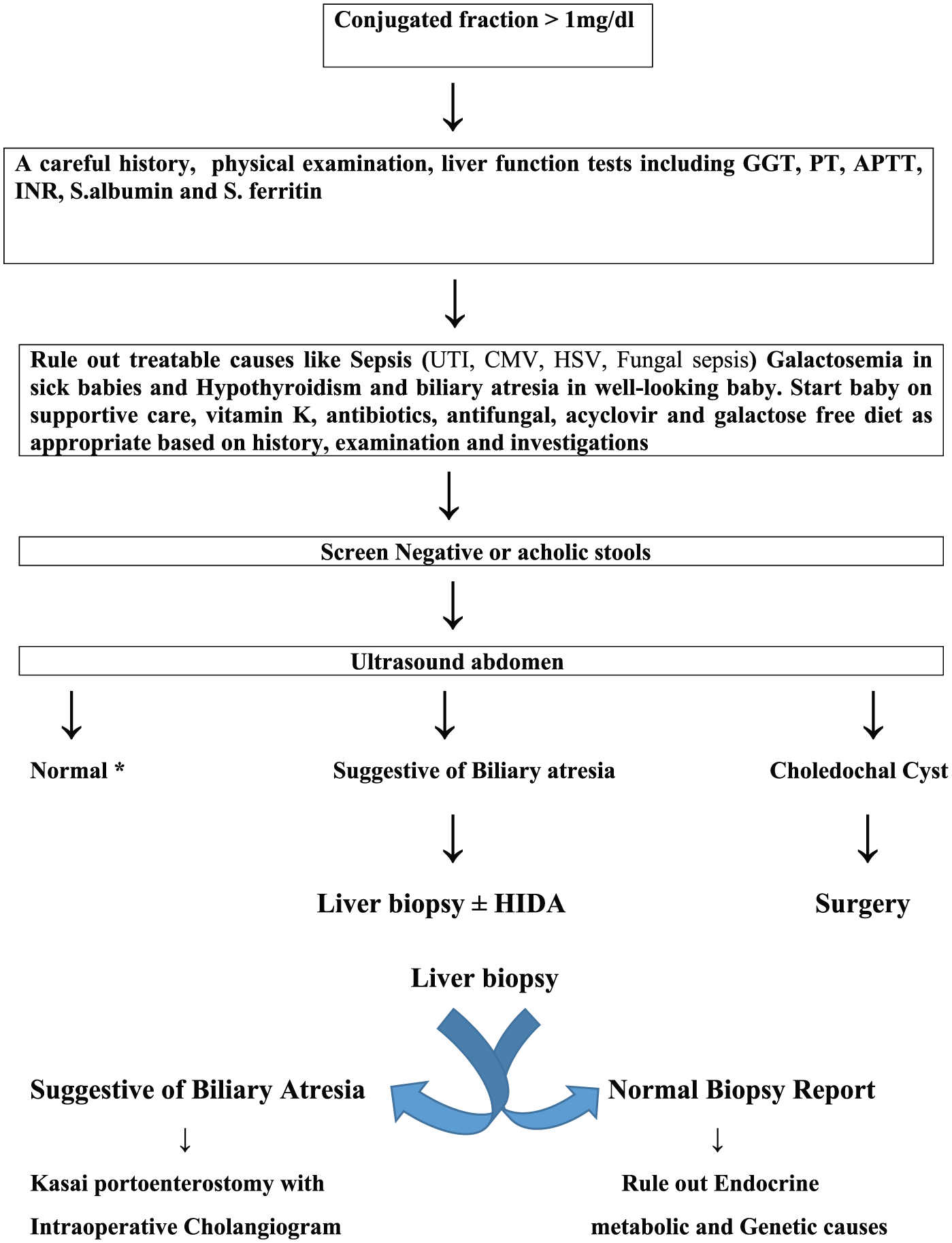
A schematic diagram to work up a patient with neonatal cholestasis syndrome.
Role of Genomics in the evaluation of NC: Genetic Cholestasis
Use of genomics especially whole-genome sequencing (WGS/WES) and next-generation sequencing has an increasing role in the diagnosis of rare disorders in ill neonates and has the potential to augment or modify the care of this vulnerable population of patients. 36 However, cost, interpretation of results and availability are the major obstacles in ordering these investigations. Nevertheless, genomics has made the biggest impact in neonatology than any other specialties of medicine. In addition, the advantage of diagnosis in antenatal period cannot be ignored. Keeping the above constrains in mind, targeted genomics is the need of the hour. Targeted genetic testing represents a multitiered approach offered either presymptomatically or postsymptomatically. Presymptomatic testing is offered to individuals with a known genetic disorder in the family, whereas postsymptomatic genetic testing should be offered to individuals suspected clinically to be affected by genetic disease, ranging from gene specific to panel specific to WGS/WES. 37 Some of the disorders which can be unveiled with genomic testing are neonatal intrahepatic cholestasis caused by citrin deficiency (mutations in SLC25A13), 38 progressive familial intrahepatic cholestasis 39 (mutations in either ATP8B1 or ABCB11), Alagille syndrome (mutations in either JAG1 or NOTCH2), Alagille syndrome and Dubin-Johnson syndrome (mutations in ABCC2). In a recent study by Togawa et al, they analyzed 109 Japanese infants with cholestasis and made a molecular genetic diagnosis in 26% of their cohort (28/109). In addition, 7.7% categorized clinically as of unknown cause were successfully categorized by the molecular genetic diagnosis. 40 In a similar report, Liu et al 41 reported the use of jaundice chip, which includes 5 disease-causing genes: SERPINA1, JAG1, ATP8B1, ABCB11, and ABCB4. With more and more research, we expect the percentage of patients diagnosed as genetic cholestasis to increase and a corresponding decrease in the unknown cause group. At present, we recommend a targeted genomic diagnosis in patients with NC especially those with atypical features.
Conclusions
Neonatal cholestasis must be considered in any infant presenting with prolonged jaundice longer than 2 weeks or early if associated with hepatomegaly, failure to thrive, acholic stools, or dark urine. Neonatal cholestasis, although rare, is a life-threatening condition unless timely diagnosed and managed appropriately. However, delay in diagnosis of NC, particularly of biliary atresia, remains a problem. Misinterpretation with physiological jaundice, lack of national-level screening programs for inborn errors of metabolism, and presence of pigmented stools are often the cause of delay in diagnosis. Universal screening for NC still has a long way to go. There is a need for strict follow-up, use of stool card, and awareness. Even if there is no specific management, appropriate supportive care and nutritional rehabilitation may be life-saving in most cases of NC.
Footnotes
Funding:
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
AP wrote the initial manuscript, VG helped in literature search, GG and AP did the critical appraisal.