
Abstract
Congenital hepatic cyst is a rare and nonsymptomatic condition in infants and children. Its incidence is 2.5% in the postnatal life with a much lower incidence in the prenatal period. Incidental finding on antenatal imaging is the most common presentation. We present a case of a newborn in whom fetal ultrasound detected a cyst within the fetal liver. Postnatal imaging revealed a liver cyst in the right lobe of the liver, with no other intrahepatic structure affected. Liver function tests were abnormal, but the patient was asymptomatic. Posterior follow-up imaging showed a minor decrease in size. Management of congenital hepatic cyst is usually conservative, done with periodic ultrasound monitoring. However, surgical treatment is the mainstay of treatment when hydrops, progressive enlargement, hemorrhage, torsion, or compression of adjacent structures occurs. Malignant transformation can occur, but it is extremely rare. Partial or total removal of the cyst is the preferred treatment in neonates with a large lesion.
Introduction
Congenital hepatic cyst is a rare and nonsymptomatic condition in infants and children. 1 Its natural history is silent and diagnosis is usually made incidentally on radiologic imaging. 2 Nonetheless, larger cysts can become symptomatic requiring surgical treatment. We present a case of congenital hepatic cyst.
Case Presentation
A 33-year-old female multigravida presented to our institution for her prenatal care. At 25 weeks of gestation, a fetal sonogram reported a 1.3 cm × 1.2 cm × 1.0 cm cyst within the fetal liver (Figure 1). At that time, it was uncertain whether it represented a choledochal etiology or an isolated parenchymal cyst. At 37 + 5 weeks of gestation, the mother was noticed to have oligohydramnios, which persisted despite the administration of intravenous fluids. She was admitted for induction of labor and a female newborn was delivered via vaginal delivery after 38 weeks of gestation. The Apgar score was 8 and 9 at 1 and 5 minutes, respectively. The physical examination of the infant soon after delivery showed a newborn girl with a birth weight of 3285 g, head circumference of 33.5 cm, chest circumference of 34 cm, abdominal circumference of 31.5 cm, and length of 51 cm. Abdominal examination revealed a soft, nondistended abdomen, with no masses palpated.

Liver cyst on prenatal ultrasound.
On the second day of life, we performed an abdominal sonogram that revealed a liver cyst in the right lobe of the liver, measuring 3.1 cm × 1.8 cm × 2.8 cm (Figure 2). The gallbladder appeared unremarkable, with no intrahepatic biliary duct dilatation (Figure 3). The hepatic function panel showed an aspartate aminotransferase level greater than 3 times the upper limit of normal (134 U/L). The infant remained asymptomatic and was discharged home with a total bilirubin level of 4.6 mg/dL, corresponding to the low-risk zone at 35 hours of life according to the Bhutani nomogram.

Liver cyst on ultrasound at day 2 of life.
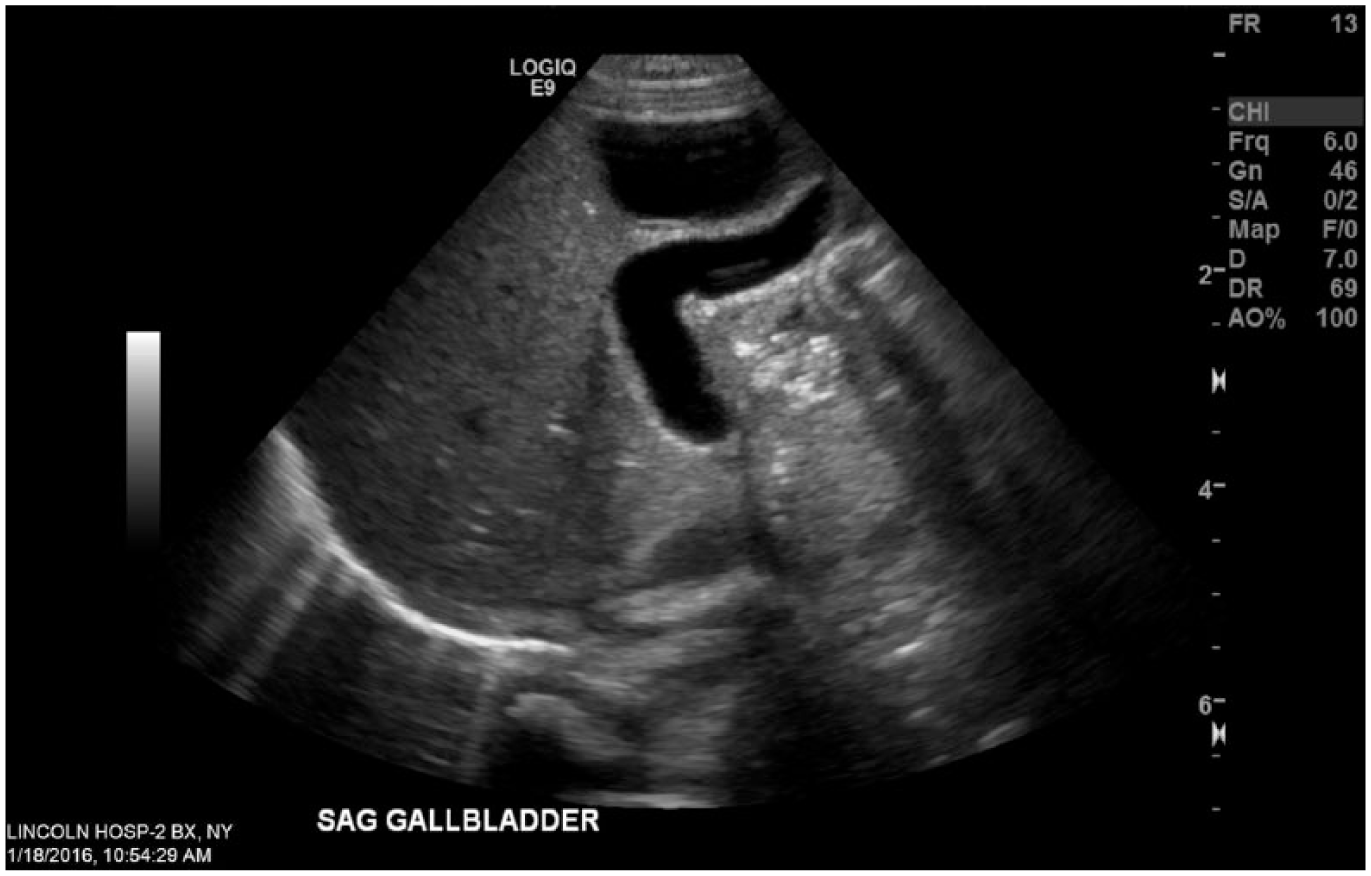
Gallbladder on ultrasound at day 2 of life.
The baby was evaluated by our pediatric gastroenterologist who recommended repeating the sonogram to ensure stability. As such, a follow-up sonogram was performed at 4 months of life showing a cyst measuring 1.9 cm × 1.1 cm × 2 cm in the right lobe of the liver with no increased vascularity. She was reevaluated by pediatric gastroenterologist at 7 months of age when a new ultrasound was performed showing complete disappearance of the simple hepatic cyst (Figure 4), with normalization of the hepatic function panel.

Resolution of liver cyst on ultrasound at 7 months of age.
Discussion
The incidence of congenital hepatic cyst in the postnatal life is 2.5%. This incidence is lower during the prenatal period with a few cases described in the literature.2,3 They are more common in girls, not associated with cysts in other organs, and rarely communicate with the biliary tree. 4 These cysts do not contain bile and arise from congenital or secondary obstruction of the biliary glands, which normally arise from the ductal plate at the hepatic hilum around the seventh week of gestation and continue to proliferate until adolescence. 1
They are superficially located just under the liver capsule and are coated by a single layer of cuboidal or columnar epithelium, characteristic of the bile ducts. The cystic fluid is generally clear and rarely contains bile. 1 The research has shown that the epithelium lining the cyst is sensitive to hormones, especially estrogen, and also that the cysts tend to enlarge in patients on hormone therapy. 5 Hepatic cysts related to these disorders may also have a genetic component, and they can appear later in life.
Incidental finding of an asymptomatic lesion on antenatal imaging is the most common presentation of a congenital hepatic cyst. In infants, symptoms consist of abdominal distension, feeding difficulties, respiratory distress, and duodenal obstruction. 4 Cholestasis may also be observed in some cases due to the compression of the hepatic parenchyma and the biliary system by a large cyst. For older patients, symptoms may include abdominal pain, nausea, and vomiting and rarely obstructive jaundice or portal hypertension. 6
The diagnosis is primarily made via radiographic studies. Ultrasonography shows the congenital hepatic cyst as an anechoic unilocular fluid-filled space with a posterior acoustic enhancement. Septations represent bridging of bile ducts and vessels and are commonly absent. Magnetic resonance imaging (MRI) typically reveals a well-demarcated water-attenuated lesion without enhancement after gadolinium with low-intensity signal on T1 and high-intensity signal on T2 images. 7 In our case, no MRI was indicated as the infant was asymptomatic and appropriate follow-up was ascertained.
The main differential diagnosis of congenital hepatic cyst includes solitary liver cyst and choledochal cyst. The former may contain bilious or nonbilious fluid and can have connection to the biliary tract. 8
Malignant transformation of a congenital hepatic cyst is extremely rare, and the only recognized risk factor for this transformation is a cyst size greater than 12 cm.4,6
Management of congenital hepatic cyst is conservative with periodic ultrasound monitoring to ensure their stability, especially for large cysts (⩾4 cm in diameter).7,9 Most simple hepatic cysts are benign in nature and have a spontaneous resolution as in our patient. 10 Surgical intervention with aspiration, sclerotherapy, or excision is indicated only for severe cases, such as hydrops, progressive enlargement, hemorrhage, torsion, or if image characteristics prompt diagnostic doubt.4,9 Compression of intrahepatic structures due to an increase in the cyst size also prompts surgery; hence, ultrasound follow-up is important.4,7 In 2 previous series published, only 2 of 12 infants with simple hepatic cysts required surgical intervention due to gradual enlargement and symptoms, and the others needed no further intervention. 4
Conclusions
Congenital hepatic cyst is a condition with a narrow differential diagnosis. It is crucial for the neonatologist to be familiar with the range of pathologies possibly causing this condition.
Accurate diagnosis is important followed by periodic surveillance with ultrasonography for optimal management of large asymptomatic lesions, as many cysts can produce obstructive biliary complications and have malignant potential.
Surgical treatment with partial or total removal of the cyst is the favored treatment in neonates with a large symptomatic lesion, with an objective of excising as much of the cyst wall as possible without causing damage to the surrounding vital organs.
Footnotes
Peer review:
Four peer reviewers contributed to the peer review report. Reviewers’ reports totaled 214 words, excluding any confidential comments to the academic editor.
Funding:
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
AR is responsible for the concept of the case report. AR wrote the first draft of the manuscript. TZ contributed to the writing of the manuscript. JG and BR reviewed and made critical revisions to the manuscript. All authors read and approved the final manuscript.