
Abstract
CD34+ hematopoietic progenitor cells have been shown to be susceptible to HIV-1 infection, possibly due to a low-level expression of CXCR4, a coreceptor for HIV-1 entry. Given these observations, we have explored the impact of forskolin on cell surface expression of CXCR4 in a cell line model (TF-1). The elevation of intracellular cyclic adenosine monophosphate (cAMP) by forskolin through adenylyl cyclase (AC) resulted in transcriptional upregulation of CXCR4 with a concomitant increase in replication of the CXCR4-utilizing HIV-1 strain IIIB. Transient expression analyses also demonstrated an increase in CXCR4-, CCR5-, and CXCR4-/CCR5-utilizing HIV-1 (LAI, YU2, and 89.6, respectively) promoter activity. Studies also implicated the protein kinase A (PKA) pathway and the downstream transcription factor CREB-1 in interfacing with cAMP response elements located in the CXCR4 and viral promoter. These observations suggest that the cAMP signaling pathway may serve as a regulator of CXCR4 levels and concomitantly of HIV-1 replication in bone marrow (BM) progenitor cells.
Introduction
In humans, active HIV-1 replication in the bone marrow (BM) has been suggested based on reports that the viral load was very similar to the viral load in the blood, in terms of both cell-free
1
and cell-associated virus.
2
Within BM cell subpopulations, CD34+ hematopoietic progenitor cells express the HIV-1 receptor CD4 and low levels of the coreceptor CXCR43 and have been reported to be susceptible to HIV-1 infection in vitro with varying efficiency.4,5 Several investigators have shown that receptor and coreceptor expression correlates with the susceptibility of progenitor cells to HIV-1.4,6–9 Therefore, it is likely that physiological events that elevate levels of CXCR4 facilitate infection of progenitor cells by increasing the net availability of the coreceptor at the cell surface. It has been reported that cyclic adenosine monophosphate (cAMP)-dependent upregulation of CXCR4 in human T-lymphocytes is mediated by a cAMP-responsive element (CRE) in the 5′ untranslated region (UTR) of its promoter.
10
Therefore, it was of interest to determine whether a similar cAMP induction would augment CXCR4 levels and HIV-1 infection in BM progenitor cells using quantitative real-time polymerase chain reaction (qRT-PCR) and in vitro viral replication assays. The specific adenylyl cyclase (AC) activator forskolin was used to augment intracellular cAMP levels in the CD34+CD4+CXCR4+ human BM progenitor cell line TF-1. Interestingly, we have previously identified a CRE sequence upstream of the transcription start site in the U3 region of the HIV-1 promoter (designated the long terminal repeat [LTR]), and this cis–acting element (TGC
Materials and methods
Reagents
Forskolin (Sigma-Aldrich), a diterpene obtained from Coleus forskohlii, was used to elevate intracellular cAMP concentrations in TF-1 cells. The PKA-specific inhibitor H-89 was also obtained from Sigma-Aldrich. Lysis buffers for nuclear extract preparation were treated with Protease Inhibitor Cocktail Set III and Phosphatase Inhibitor Cocktail Set II, which were obtained from Calbiochem. Total cellular RNA for TaqMan qRT-PCR was prepared from TF-1 cells using TRI Reagent (Sigma-Aldrich).
Cell culture and treatment procedures
The TF-1 CD34+CD38+ cell line (American Type Culture Collection [ATCC]) was grown in Roswell Park Memorial Institute (RPMI) 1640 medium (ATCC) supplemented with 10% heat-inactivated fetal bovine serum (HyClone), penicillin (100 U/mL), streptomycin (100 µg/mL), and recombinant human granulocyte/macrophage colony-stimulating factor (2 ng/mL; eBioscience) and maintained at 37°C in 5% CO2 at 90% relative humidity.
Cloning and site-directed mutagenesis
The HIV-1 LAI, YU2, and 89.6 LTRs were cloned into the pGL3-Basic vector (Promega) with the firefly luciferase (luc) gene. The LAI-LTR-Luc, YU2-LTR-Luc, and 89.6 LTR-Luc reporter constructs have been described previously.13,14 Site-directed mutagenesis was performed using the QuikChange mutagenesis procedure (Stratagene) on all three HIV-1 LTR-Luc reporter vectors to incorporate a TG mutation in the ATF/CREB site that is reported to exhibit a knockout phenotype with respect to binding CREB-1 (Santa Cruz Biotechnologies). The following primers were used for site-directed mutagenesis: LAI-LTR (5′-CAA-GAA-CTG-CTG-TGA-TCG-AGC-TTG-CT-3′), YU2-LTR (5′-CAA-GAA-CTG-ATG-TGC-TCG-AGC-TTT-CT-3′), and 89.6 LTR (5′-ACT-GAA-CTG-CTG-TGA-CTG-AGC-TAT-CT-3′). Sequences were confirmed and analyzed using Lasergene software (DNASTAR, Inc.).
Transient transfections and expression analyses
TF-1 cells (2 × 106) were transiently transfected with the HIV-1 parental or mutated TG firefly luciferase-LTR constructs (1 µg) in conjunction with the pRL-TK Renilla luciferase (Promega) internal control vector (50 ng) or a combination of wild-type luciferase-LTR constructs, pRL-TK, and CREB-133/KCREB vectors (Clontech Laboratories, Inc.) using the Amaxa Nucleofector System T procedure (Amaxa). After transfection, cells were placed in new media and allowed to recover for an hour. Cells were then either left untreated for 24 hours or pretreated with H-89 (10 µ
Isolation of RNA and qRT-PCR
TF-1 cells were serum starved for 1 hour and subsequently total RNA was isolated from forskolin (100 µ
Nuclear extract preparation and EMS assays
Large-scale nuclear extracts were prepared from forskolin-treated (100 µ
HIV-1 p24 ELISA
Cells were seeded into 12-well plates at a density of 0.5 × 106 cells/mL and incubated for 24 hours in the absence or presence of forskolin (100 µ
Results
Identification of CREs in the HIV-1 LTR and CXCR4 5′ UTR
The LAI, YU2, and 89.6 HIV-1 LTR sequences were obtained from the Los Alamos Sequence Database (GenBank accession numbers X01762, M93258.1, and U39362.2, respectively). The CXCR4 promoter-5′ UTR sequence was previously published (GenBank accession ID AJ224869.1). 16 Analysis of these sequences using the online transcription factor binding site software Match from the TRANSFAC 7 public database (www.gene-regulation.com) identified the presence of a single CRE in the entire CXCR4 promoter-5′ UTR (+23 to +30) and in the HIV-1 LTR located upstream of the transcription start site (−118 to −127) by adopting a stringent cutoff ratio (matrix similarity = 0.8, core similarity = 0.85) (Figure 1A).

Forskolin induces transcriptional activation of HIV-1 LTR and upregulation of CXCR4. (A) The molecular sequence of CREs in the LTR of reference HIV-1 strains and in the 5′ UTR of the CXCR4 gene. TRANSFAC analysis identifies the presence of a lone CRE in the CXCR4 promoter-5′ UTR and the HIV-1 LTRs. The sequences are compared with a classical consensus CRE in the T-lymphocyte SH2D2A promoter, and the variant base pairs are marked in red. (B) With forskolin stimulation (the range of concentrations included 0, 10, 25, 50, 75, and 100 µ
Effects of intracellular cAMP augmentation on LTR-directed transcription
Forskolin, a diterpene obtained from C. forskohlii, is a potent and specific activator of AC and was used to elevate intracellular cAMP levels in TF-1 cells. Transient transfection assays were utilized to measure LTR-driven firefly luciferase gene expression as a direct measure of HIV-1 LTR activation. Titration of transfected TF-1 cells with increasing concentrations of forskolin exhibited a dose-dependent increase in HIV-1 LAI, YU2, and 89.6 LTR activity. After the results were normalized to Renilla luciferase expression and plotted as fold over untreated samples, it was apparent that the HIV-1 LAI-LTR was the most inducible (6.2-fold), the YU2-LTR was intermediate (2.9-fold), and the 89.6 LTR was the least (1.9-fold) (Figure 1B). This differential responsiveness was proposed to be a reflection of the variation in the genetic sequence of the respective CREs specifically and the LTR in general.
Site-directed mutagenesis of the HIV-1 LAI-, YU2-, and 89.6 LTR-luc constructs to incorporate an AC to TG mutation in the first half of the CRE site in conjunction with transient transfections exhibited an abrogation in activity of these LTRs in response to forskolin. After the results were normalized to Renilla luciferase expression and compared with basal LTR activity, the fold change of 100 µ
Kinetics of cAMP-induced CXCR4 transcription in TF-1 cells
qRT-PCR assays were performed to study the regulation of CXCR4 transcription in response to enhanced cAMP levels (Figure 1D). Kinetics revealed a biphasic curve, wherein transcript levels always remained elevated and returned close to the untreated threshold level only at 24 hours after forskolin stimulation. The amplitude of the curve was observed at 6–9 hours (4-fold increase), with transcript levels declining at 15 hours (1.92-fold). Subsequently, a smaller peak was observed at 18 hours (2.8-fold). These results suggest functional significance of the CRE with respect to mediating cAMP-induced CXCR4 transcription. The biphasic nature of the response to forskolin is a likely outcome of the kinetics and dynamics relative to the interaction of cognate transcription factors with the LTR-specific CRE.
Binding of CREB-1 to identified CREs in vitro in TF-1 cells
To analyze the molecular basis of cAMP-induced transcription mediated by the CREs in HIV-1 and CXCR4 promoter/UTR, EMS assays were performed to detect the presence of the transcription factor CREB-1 in complexes formed between forskolin-stimulated TF-1 nuclear extracts and radiolabeled parental and TG-mutated oligonucleotides (Figure 2). Compared with the “probe-only” reaction (lane 1), a specific DNA–protein complex was detected in the case of (A) LAI, (B) YU2, (C) 89.6, and (D) CXCR4 parental CRE probes (lane 2). This complex consisted of three distinct bands (asterisk) that exhibited either abrogation (Figure 2B and C) or a combination of both abrogation and supershift (Figure 2A and D) when the reaction was incubated with a CREB-1-specific antibody (lane 4). Addition of an isotype antibody did not affect the complex (lane 3). The upper two bands likely represent the 43-kDa ubiquitous phosphorylated CREB-1-α,Δ activator isoforms, 12 whereas the lower band is most likely the homologous family member phosphorylated ATF-1. Incubation with mutant TG oligonucleotides prevented the formation of the CREB-1 complex, indicating the likely involvement of CREB-1 in binding and activation of the identified CREs.

CREB-1 is present in the DNA–protein complexes formed between forskolin-stimulated TF-1 nuclear extracts and identified CREs. EMS assays performed with forskolin-stimulated TF-1 nuclear extracts and radiolabeled probes corresponding to (A) LAI, (B) YU2, (C) 89.6, and (D) CXCR4 parental CREs revealed the presence of CREB-1 43-kDa protein in the DNA–protein complex (asterisk). Although the complex was unaffected by an isotype antibody, it was abrogated/supershifted with a CREB-1-specific antibody. Corresponding reactions with mutant TG CRE radiolabeled oligonucleotides exhibit the absence of the CREB-1-containing complex, suggesting that CREB-1 directly binds to parental CREs in vitro. Two other DNA–protein complexes (double and triple asterisks) are also present and are nonspecific in nature as they remain unaffected by addition of specific antibody.
Involvement of CREB-1 in CXCR4 and HIV-1 LTR-directed transcription in TF-1 cells
Transient transfection assays with dominant-negative CREB-1 constructs were employed to definitively implicate CREB-1 in cAMP-induced HIV-1 LTR activation. The KCREB construct (pKCREB) encodes a form of CREB-1 with mutations in its DNA-binding domain. The inability to bind CRE results in mutant CREB-1 acting as a dominant-negative repressor, blocking cAMP-induced transcription. The CREB-133 construct (pCREB-133) also encodes a mutated form of CREB, preventing Ser133 CREB-1 phosphorylation required for transactivation, thereby blocking cAMP-induced transcription.
In comparison with forskolin-stimulated cells transfected with parental LTR-luc constructs and empty vector, cells transfected with LTR-luc and CREB-133 or KCREB constructs exhibited diminution in firefly luciferase expression (Figure 3A). In the case of LAI, absolute expression decreased from 1 (arbitrarily set for LAI-LTR-luc) to 0.27 (LAI-LTR-luc + KCREB) and 0.32 (LAI-LTR-luc + CREB-133). In the case of YU2, absolute expression decreased from 1 (arbitrarily set for YU2-LTR-luc) to 0.68 (YU2-LTR-luc + KCREB) and 0.56 (YU2-LTR-luc + CREB-133). In the case of 89.6, absolute expression decreased from 1 (arbitrarily set for 89.6 LTR-luc) to 0.38 (89.6 LTR-luc + KCREB) and 0.54 (89.6 LTR-luc + CREB-133). As observed in Figure 1 with TG-mutated CREs, the LAI-LTR is the most susceptible LTR with respect to inhibition by the dominant-negative CREB-1. However, in contrast to earlier observations, it is unclear why YU2 is less susceptible to dominant-negative CREB-1 in comparison with 89.6. In the case of LAI, the difference may be partly explained by an absolute requirement for CREB-1-CRE complex formation. We speculate that, in the case of YU2 and 89.6, other transcription factors and sequences adjacent to the HIV-1 LTR CRE may also play a role in facilitating cAMP-induced LTR activation.
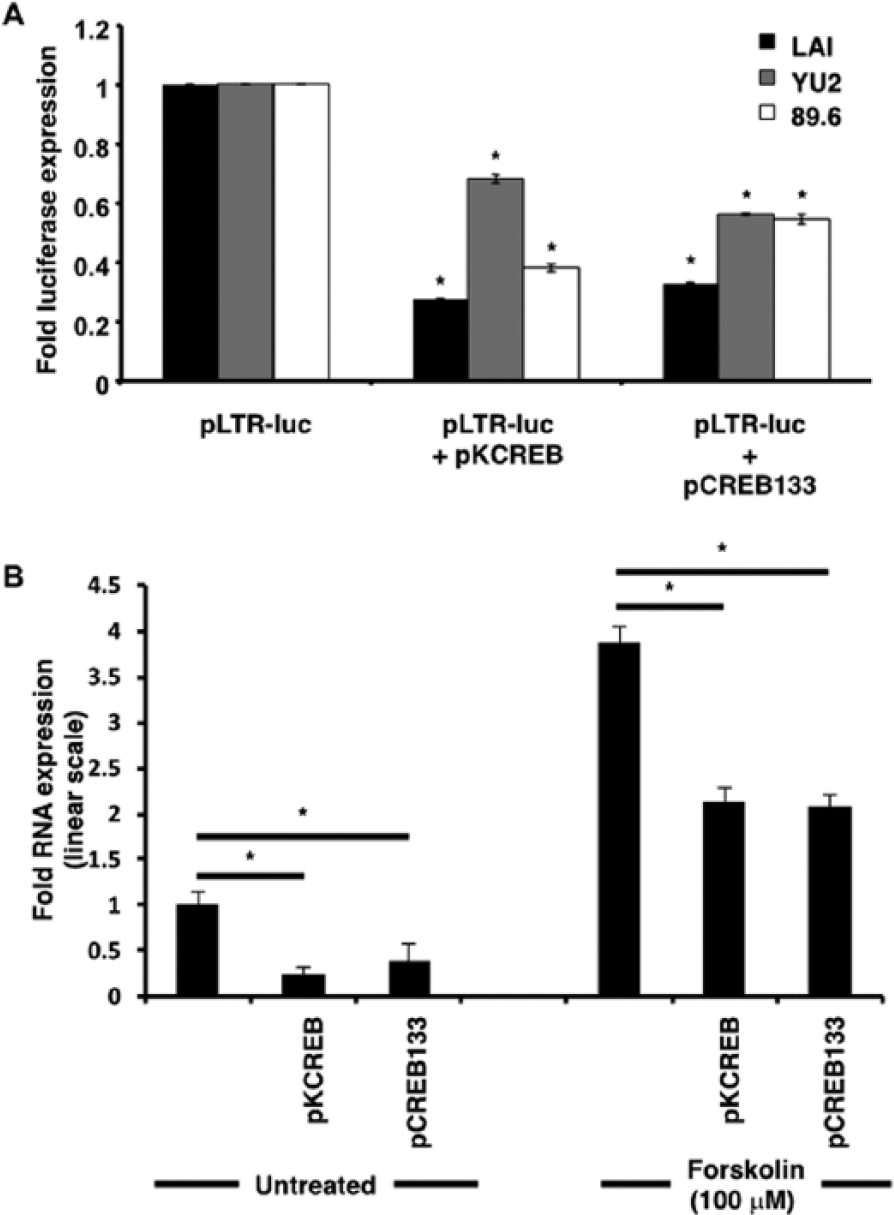
cAMP-induced transcriptional activation of HIV-1 and CXCR4 promoters is specifically mediated by CREB-1. (A) In comparison with forskolin-stimulated cells transfected with parental LTR-luc constructs alone, cells with LTR-luc and CREB-133 or KCREB constructs exhibit diminution in firefly luciferase expression. All firefly luciferase readings were normalized to Renilla readings by cotransfecting cells with the pRL-TK Renilla luciferase construct and expressed as ratios of forskolin-treated to untreated samples. (B) qRT-PCR assays revealed that in comparison with untransfected untreated and untransfected forskolin-treated samples, pKCREB and pCREB-133 transfected samples showed diminution in CXCR4 transcript levels. Results were normalized to RPLPO expression and expressed in linear scale as fold over untreated untransfected samples. Asterisks indicate significance of P < 0.05 using Student’s t–test.
qRT-PCR assays with dominant-negative CREB-1 constructs were similarly used to reinforce the involvement of CREB-1 in CXCR4 transcription. In comparison with untransfected forskolin-treated samples, both pKCREB and pCREB-133 transfected samples showed 2-fold decline in levels of CXCR4 mRNA at nine hours after forskolin treatment. In the case of untransfected untreated samples, this decline was a little more pronounced (Figure 3B). As expected, transcription was not suppressed completely due to the presence of endogenous CREB-1 and other transcription factor isoforms belonging to the ATF/CREB family in TF-1 cells.
Effects of PKA inhibition on cAMP-induced CXCR4 and HIV-1 LTR-directed transcription
We have previously identified PKA as a primary facilitator of CREB-1 phosphorylation in TF-1 cells in response to cAMP augmentation. 17 Therefore, to implicate PKA as a key intermediary in the linear pathway from cAMP to CREB-1, transient transfections and qRT-PCR experiments were performed, assaying for firefly luciferase expression and CXCR4 mRNA levels, respectively, in the absence or presence of the PKA inhibitor H-89.
The forskolin-stimulated HIV-1 LAI-, YU2-, and 89.6 LTR-luc expression levels were consistent with those observed in Figure 1B. However, the result obtained with the 89.6 LTR was not shown to be statistically significant due to the larger error in this experiment. In the presence of H-89 alone, there was a marginal reduction, although not statistically significant, with respect to basal LTR-luc levels. This is probably due to the fact that CREB normally plays a role in transactivating the LTR under basal conditions. So inhibiting PKA from phosphorylating CREB under basal conditions can and probably should still have some effect. In the case of forskolin stimulation, H-89 pretreatment completely abrogated luciferase expression from all three HIV-1 promoters (Figure 4A). This result confirmed the presence of PKA as the immediate effector of cAMP and suggested that PKA inhibition obliterates, in part, downstream activation of CREB-1, thereby abolishing cAMP-induced transcription.

Increased cAMP-induced expression from HIV-1 and CXCR4 promoters is facilitated by PKA. (A) In comparison with untreated samples, forskolin-stimulated cells exhibit up to a ~6-fold upregulation in luciferase expression. This upregulation is completely inhibited by H-89 pretreatment for one hour. H-89 alone slightly diminishes basal luciferase expression. All firefly luciferase readings were normalized to Renilla readings by cotransfecting cells with the pRL-TK Renilla luciferase construct and expressed as fold over untreated samples. (B) qRT-PCR assays revealed that in comparison with untreated samples, forskolin-treated samples showed a ~4.6-fold increase in CXCR4 transcript levels. This was reduced by 50% when samples are pretreated with H-89. Results were normalized to RPLPO expression and expressed in linear scale as fold over untreated samples. Asterisks indicate significance of P < 0.05 using Student’s t–test.
Similarly, forskolin-stimulated CXCR4 mRNA levels were consistent with those observed in Figure 1D. In comparison with untreated samples, there was a maximum upregulation at nine hours (4.6-fold). After pretreatment with H-89, this upregulation was inhibited by 50% (2.3-fold). H-89 treatment alone did not significantly affect basal CXCR4 mRNA levels (Figure 4B). In contrast to HIV-1 LTR activity, this suggested only partial dependence of CXCR4 transcription on PKA. This result was consistent with our previous experiments wherein CREB-1 phosphorylation was not completely abolished after PKA inhibition, 17 indicating the likely role of other kinases in mediating this effect.
Effects of cAMP augmentation on HIV-1 replication in TF-1 cells
To understand if forskolin upregulation of CXCR4 in TF-1 cells resulted in increased virus infection, viral infectivity assays were performed using a CXCR4-utilizing HIV-1 strain IIIB. These experiments revealed an increase in replication (3.3-fold) of the HIV-1 strain IIIB in response to forskolin treatment (Figure 5). Along with the results obtained from LTR activation experiments, these results suggested that increased CXCR4 levels might translate into increased viral entry and subsequent replication based on the observed increase in LTR-directed transcription. Additional experiments will assess the presence of unspliced and multiple spliced transcripts to confirm this observation.

Forskolin treatment enhances replication of the CXCR4-utilizing HIV-1 strain IIIB in TF-1 cells. Assays for HIV-1 p24 were performed on cellular supernatants 24 hours after infection to assess HIV-1 IIIB replication in response to 100 µ
Discussion
Because HIV-1 has been reported to be present in the BM of HIV-1-infected patients, it is likely that CD34+CD4+CXCR4+ BM progenitor cells are susceptible to infection by at least a subset of HIV-1 viral strains. However, BM stromal cells produce in vivo large quantities of stromal cell-derived factor-1α (SDF-1α), a member of the family of α-chemokines and the only natural and endogenous ligand of CXCR4. 18 CD34+CD38+ BM progenitors have also been reported to be positive for SDF-1α. 3 More importantly, SDF-1α competes with CXCR4-utilizing HIV-1 with respect to binding to the coreceptor CXCR4, 19 blocking HIV-1 entry, 18 and productive infection. 20 Therefore, progenitors are assumed to be partially refractile to infection in vivo. In this scenario, competition for occupation of CXCR4 by HIV-1 can be modulated by either a decline in SDF-1α production or an augmentation in levels of the coreceptor itself. Because cAMP has been reported to increase CXCR4 transcript levels in T-lymphocytes owing to the presence of a CRE, 10 it was of interest to determine whether a similar phenomenon would apply to BM progenitor cells. This would then relate to the potential of BM progenitor cells being more susceptible to HIV-1 due to the increased CXCR4 expression on the cells. Secondarily once integrated in the cell cAMP could also induce the LTR compounding the infection by driving more viral transcription and hence replication. The in vivo significance of this question is not lost as patients with late-stage HIV-1 infection exhibit very high circulating levels of prostaglandin E2, 21 a natural cAMP-inducing ligand. For our studies, the well-characterized CD4+CXCR4+ human BM progenitor cell line TF-1 and the cAMP-enhancing AC-activator forskolin were selected for use in the experimental design. A biphasic wave of induction was observed in CXCR4 transcription in TF-1 cells. We also determined whether elevated cAMP levels would affect HIV-1 promoter activity, given that we have previously identified a CRE in the HIV-1 LTR. 11 The presence of a single CRE in the LTRs of LAI, YU2, and 89.6 strains was detected by TRANSFAC analyses by comparison with a consensus CRE. In addition to the structural differences among the CREs in the LTRs of the individual HIV-1 strains, these elements were observed to be different from the CRE present in the CXCR4 5′ UTR. These genetic alterations are likely to affect the responsiveness of these genes to a cAMP stimulus due to the potential for differential recruitment of cAMP-responsive transcription factors to CREs with different sequence variants. This possibility was reiterated in transient transfection assays wherein the LAI-LTR was found to be most inducible in comparison with the YU2 and 89.6 promoters. Moreover, the cAMP response was found to be directly mediated by the identified CRE as apparent from site-directed mutagenesis studies. qRT-PCR and transfection analyses also identified PKA and the transcription factor CREB-1 as key sequential effectors of this dual-pronged stimulation of CXCR4 and HIV-1 LTR-mediated transcription. Finally, an observed increase in replication of CXCR4-utilizing IIIB strain in forskolin-stimulated TF-1 cells highlights the utility of these cells as a physiologically relevant cellular model (Figure 6) for determining the effects of intracellular cAMP elevation on HIV-1 infection as well as gene expression in progenitor cells.

The cAMP-PKA-CREB-1 pathway in TF-1 cells. Forskolin treatment enhanced intracellular cAMP levels by directly activating membrane-bound AC. This resulted in activation of the PKA catalytic C subunits, which diffuse into the nucleus. Once inside, they phosphorylated CREB-1 activator isoforms. Activated CREB-1 binds to CREs in the HIV-1 and CXCR4 promoter/5′ UTRs, thereby increasing transcription. This may have resulted in increased surface levels of CXCR4 and an increase in HIV-1 susceptibility.
Footnotes
Acknowledgements
We thank Adriano Ferruci for his technical assistance with the transient transfections.
Peer Review:
Five peer reviewers contributed to the peer review report. Reviewers’ reports totaled 965 words, excluding any confidential comments to the academic editor.
Funding:
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: These studies were funded in part by the Public Health Service, National Institutes of Health, through grants from the National Institute of Neurological Disorders and Stroke (NS32092 and NS46263, Dr. Brian Wigdahl, Principal Investigator; NS089435, Dr. Michael R. Nonnemacher, Principal Investigator), the National Institute of Drug Abuse (DA19807, Dr. Brian Wigdahl, Principal Investigator), and National Institute of Mental Health Comprehensive NeuroAIDS Center (CNAC) (P30 MH092177, Kamel Khalili, PI; Brian Wigdahl, PI of the Drexel subcontract). Drs. Michael Nonnemacher and Fred Krebs were also supported by faculty development funds provided by the Department of Microbiology and Immunology and the Institute for Molecular Medicine and Infectious Disease. The authors confirm that the funder had no influence over the study design, content of the article, or selection of this journal.
Declaration of Conflicting Interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author contributions
Conceived and designed the experiments: AB, LL, VP, BW, and MRN. Analyzed the data: AB, LL, VP, BW, and MRN. Wrote the first draft of the manuscript: AB. Contributed to the writing of the manuscript: AB, LL, VP, FCK, BW, and MRN. Agreed with manuscript results and conclusions: AB, LL, VP, FCK, BW, and MRN. Jointly developed the structure and arguments for the paper: AB, LL, VP, FCK, BW, and MRN. Made critical revisions and approved final version: AB, LL, VP, FCK, BW, and MRN. All the authors reviewed and approved the final manuscript.