
Abstract
Objective:
The current study aimed to investigate the role of β and α1-adrenoceptors in pancreatic regulation of glucose homeostasis, apoptosis, and fibrosis in a rat model of dexamethasone-induced insulin resistance.
Introduction:
Insulin resistance is a hallmark of metabolic syndrome and is often linked to glucocorticoid excess. β- and α1-adrenoceptors are key modulators of glucose metabolism and tissue remodeling, yet their roles in pancreatic dysfunction under metabolic stress remain incompletely understood. Emerging evidence highlights β-arrestin2 as a key mediator of apoptosis and fibrosis beyond classical G protein signaling.
Methods:
Insulin resistance was induced in rats by subcutaneous dexamethasone (10 mg/kg/day) for 7 days. The therapeutic effects of carvedilol, phenylephrine, phenylephrine + carvedilol, propranolol, doxazosin, and doxazosin + propranolol were evaluated in relation to glucose homeostasis and pancreatic apoptotic/fibrotic signaling.
Results and Conclusion:
Dexamethasone impaired glucose homeostasis, expanded islet mass, and triggered pancreatic cell apoptosis and fibrosis via upregulated BAX/Bcl-2 expression ratio, downregulated cAMP, upregulated PKA, ERK1/2, and CREB expression with elevated PKB activity and reduced FOXO1 expression. % Level of β-arrestin2 was reduced in pancreatic islets and elevated in exocrine pancreas in relation to the aforementioned modulations. Blockade of β- or α1-adrenoceptors significantly ameliorated these effects, with combined blockade yielding superior benefits. Carvedilol’s effects were largely β-mediated, with minor α1 involvement. Low-dose phenylephrine yielded modest improvement, supporting a context-dependent, protective role of α1-adrenoceptor activation during metabolic stress. The dependence of drugs’ effects on β-arrestin2 highlights its potential as a central regulator of pancreatic remodeling and a promising target in IR-linked pathologies.
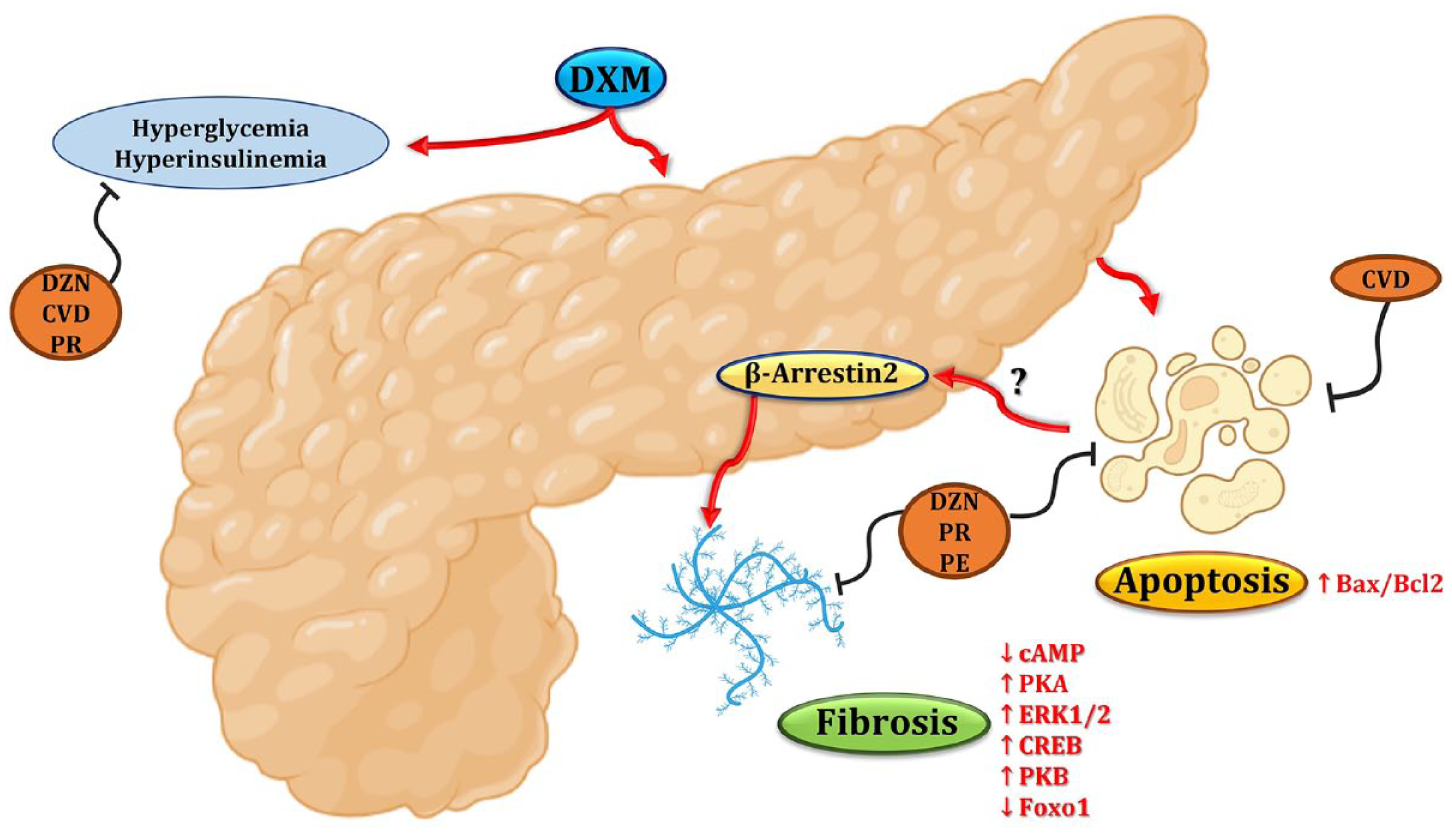
Highlights
DXM disrupted glucose homeostasis and triggered pancreatic apoptosis and fibrosis.
β- or α1-blockade reversed DXM effects; combined blockade showed superior benefits.
α1-Agonism produced modest, context-dependent improvements.
cAMP, PKA-ERK1/2-CREB, and PKB-FOXO1 modulations mediated the reported effects.
CVD, PE, PR, and DZN effects were β-arrestin2-dependent.
Introduction
Glucocorticoids, such as the here investigated dexamethasone (DXM), are highly effective immunosuppressant and anti-inflammatory drugs. They have long been used in various conditions, for example, auto-immune diseases, allergic reactions, and cancer, immensely improving quality of life and prognosis. 1 Glucose intolerance and insulin resistance (IR) are concerns in patients prescribed glucocorticoid-based therapy. 2 An insight into pancreatic β-cell adaptation to the glucocorticoid-induced IR has been introduced. Increased pancreatic islet volume due to cell proliferation and increased insulin secretion capacity were found in animals treated with glucocorticoids such as corticosterone and dexamethasone.3–5 Chronic β-cell exposure to glucocorticoids such as prednisolone was, however, associated with apoptosis and defective insulin biosynthesis. 6 In multiple organ systems, apoptosis has been identified as the initiator of the fibrotic response. 7
Understanding the network of cell signaling pathways in the pancreas may help develop novel strategies to promote cell survival. 8 A crosstalk exists between the signaling pathways of cyclic adenosine monophosphate (cAMP), protein kinase A (PKA), and extracellular signal-regulated kinases 1 and 2 (ERK1/2). cAMP induces activation of PKA, which acts upstream of mitogen-activated protein kinase kinases 1 and 2, mediating ERK1/2 phosphorylation.9,10 ERK1/2 cooperate with PKA and are responsible for half of the PKA-mediated cAMP-responsive element-binding protein (CREB) phosphorylation. 11 CREB is required for cell survival by regulating B-cell lymphoma-2 (Bcl-2) gene expression.12,13
In the same context, the regulation and function of forkhead box O (FOXO) proteins—evolutionarily conserved downstream targets of protein kinase B (PKB)—remain largely uncharacterized. 14 Sensing the balance between intracellular stress signals and growth-promoting extracellular signals, FOXO factors orchestrate the appropriate cellular responses by either apoptosis or cell growth and resistance to stress.15,16
Classically, the G protein-coupled β-adrenoceptors, in response to stress hormones, activate the adenylyl cyclase via Gsα, elevating the second messenger, cAMP, which then activates PKA. 17 Arrestins act as a regulator of cAMP-related signaling since they work together with G protein-coupled receptor (GPCR) kinases to internalize the GPCRs, such as the β-adrenoceptor. 18 It is now appreciated that β-arrestins act as signal transducers in their own right, stimulating a distinct array of signaling and cellular responses, such as ERK induction. 19 β-arrestin2 is also implicated in PKB signaling, being associated with and necessary for PKB activation in some systems. 20
Due to widespread distribution of β-adrenoceptors in the body, related signaling pathways have been thoroughly investigated, making β-adrenoceptors appreciated drug targets to design agonists and antagonists to regulate alternative signaling pathways with intervention against clinical diseases. 21 Interestingly, there exists an interaction between β and α1-adrenergic pathways, balancing insulin secretory function and/or viability requirements.22,23
Being not explicitly reported in the literature, the current study was conducted to investigate the role of β and α1-adrenoceptors in pancreatic regulation of glucose homeostasis, apoptosis, and fibrosis in DXM-treated rats as a model of IR. A new insight has been introduced into β-arrestin2 crosstalk with cAMP, PKA-ERK1/2-CREB, and PKB-FOXO1 signaling pathways as potential therapeutic targets to mitigate the DXM-induced pancreatic cell apoptosis and fibrosis.
Materials and methods
Sample size calculation
The sample size was calculated at the beginning of the experiment using the Resource Equation Approach, 24 which is commonly applied in animal studies involving group comparisons. Based on this approach, the acceptable range of degrees of freedom for the error term in analysis of variance (ANOVA) is between 10 and 20. For one-way ANOVA, the minimum and maximum sample sizes per group are calculated as 10/k + 1 and 20/k + 1, respectively, where k is the number of groups. Given that the study involved eight experimental groups, the minimum required sample size per group was three rats, and the maximum was four rats. To account for anticipated mortality associated with high-dose DXM administration, nine rats were initially assigned to each group following the approval of the Institutional Animal Care and Use Committee (IACUC) of Zagazig University (Approval No. ZU-IACUC/3/F/210/2019). All surviving animals were included in the final analyses. No animals were excluded based on experimental outcomes; only mortality led to group size reduction.
Experimental animals
In the current study, 72 adult male Wistar rats (180 ± 20 g, 8 weeks old) were obtained from the Faculty of Veterinary Medicine, Zagazig University (Egypt). Under a 12-h/12-h light/dark cycle (light on from 7 am to 7 pm and off from 7 pm to 7 am) with ad libitum access to tap water and standard pellet chow (United Feeds Co., First Industrial Zone, Obour City, Kaliobeya, Egypt), the animals were housed in plastic cages with wood shave bedding in the animal care unit at the Faculty of Pharmacy, Zagazig University. Constant temperature (23 ± 2°C) and humidity (60% ± 10%) were ensured in the animal house during the experiments. Prior to beginning the experimental work, the animals were acclimatized for 2 weeks to reduce stress and ensure they were comfortable in their environment. The animals were gently handled by trained personnel and monitored daily for distress.
Drugs and chemicals
DXM, propranolol, and doxazosin were provided by EPICO Co. (10th of Ramadan, Egypt). Carvedilol was supplied by Multi-Apex Pharmaceutical Co. (Cairo, Egypt). Phenylephrine and dimethyl sulfoxide (DMSO) were purchased from Sigma-Aldrich (St. Louis, MO, USA). All other chemicals used were of analytical grade.
Experimental design
After being acclimatized for 2 weeks, healthy rats were randomly divided into 8 experimental groups (9 rats each). Group 1 rats were intraperitoneally administered vehicle (DMSO: tween 80: water in a volume ratio of 1:1:8) and subcutaneously administered distilled water for 7 days 25 and served as the control group. Group 2 (DXM) was administered DXM dissolved in distilled water (10 mg/kg/day, S.C.) for 7 days 26 and intraperitoneally received the vehicle. Two and a half hours before DXM administration, groups 3–8 were treated with the respective agents as shown in the diagram in Figure 1. Group 3 (CVD) received carvedilol dissolved in the vehicle (10 mg/kg/day, I.P.). 27 Group 4 (PE) was administered phenylephrine dissolved in the vehicle (1 mg/kg/day, I.P.). 28 Group 5 (PE + CVD) was given phenylephrine followed by carvedilol 30 min later. Group 6 (PR) was treated with propranolol dissolved in the vehicle (30 mg/kg/day, I.P.). 29 Group 7 (DZN) received doxazosin dissolved in the vehicle (5 mg/kg/day, I.P.). 30 Group 8 (DZN + PR) was given doxazosin followed by propranolol 30 min later. All drugs were administered at literature-supported doses known to exert receptor-modulating effects in rats, allowing for a meaningful comparison of their biological effects under glucocorticoid-induced metabolic stress. The injection volume was 500 μL per 200 g body weight (BW) for all groups.

Timeline of drugs and vehicle administration.
Measurement of body anthropometric parameters
Body weights in (g) were recorded daily over the study period, using a digital analytical weighing balance. At the end of the study period and before euthanasia, tibial lengths expressed in (cm) were measured using a measuring tape, and BW to tibial length ratios in (g/cm) were calculated. Likewise, lengths and waist circumferences (WCs) were determined, beginning from the nose to the anus and around the waist at the hip region above the iliac crest; respectively. The body mass index (BMI), expressed as BW(g)/length square (cm2), was calculated.31,32
Blood and tissue sampling
At the end of the study period, the animals were subjected to a 12-h fast. The fasting range between the first and last rat euthanized was about 2 h. Rats were euthanized by decapitation. This method was approved by the IACUC and has been widely used in other studies due to its efficiency and rapid onset of unconsciousness. 33 Trunk blood was collected from the site of decapitation. Blood samples were allowed to clot for 30 min, centrifuged at 4000 rpm at 4°C for 15 min, and stored at −20°C for further analyses. Pancreas tissue samples were harvested and cut into two parts on ice packs. One part was immediately snap frozen in liquid nitrogen and stored at −80°C for later analyses. The other part was, however, fixed using 10% neutral buffered formaldehyde for histopathological and immunohistochemical examination and determination of collagen deposition.
Sample pooling
Samples from each group were randomly combined into three pools. For groups with nine animals, equal amounts from three animal samples were mixed to form each pool. In case the number of samples was seven, two pools were prepared by mixing equal amounts of each pair of samples together, and the third pool was rather prepared by mixing equal amounts of three samples. In case the number of samples was six, the samples were pooled by mixing equal amounts of each pair of samples together. In case the number of samples was 4, only two samples were pooled together, and the remaining two samples were kept separate. According to the previous statistical studies, sample pooling is a means to obtain reliable group-level estimates when the individual analysis is not feasible, and it gives more representative mean values than the random selection of a few samples for the measurement of biochemical parameters. 34
Biochemical assessment of glucose homeostasis
Fasting serum glucose (FSG) levels were measured colorimetrically using the BioMed-Glucose LifeSpan assay kit, Badr City, Egypt. 35 Fasting serum insulin (FSI) levels were determined using a rat enzyme-linked immunosorbent assay (ELISA) kit, Crystal Chem, IL, USA, 90010. Homeostasis Model Assessment of Insulin Resistance (HOMA-IR) was calculated as FSI (µU/mL) × FSG (mg/dL)/405. 36
Biochemical assessment of apoptotic and profibrotic signaling pathways in pancreatic tissue
In pancreatic tissue homogenates, Bcl-2-Associated X Protein (BAX) and Bcl-2 expression levels were quantitatively determined using rat-specific sandwich ELISA kits, LifeSpan BioSciences, Seattle, WA, USA, LS-F21494 and Elabscience, Houston, TX 77079, USA, E-EL-R0096, respectively. Expression levels of cAMP, ERK1/2, and CREB were quantitatively determined using the rat-specific sandwich ELISA kits, MyBioSource, San Diego, CA, MBS018906, Assay Genie, Dublin 1, Ireland, RTFI00754, and MyBioSource, San Diego, CA, MBS2504589, respectively. Rat-specific competitive ELISA kit, MyBioSource, San Diego, CA, MBS728647, was, however, used for quantitative determination of PKA expression levels.
Quantitative measurement of PKB activity involved a single-site, semi-quantitative immunoassay utilizing the PKB kinase inhibitor screening assay kit, Creative Diagnostics, 45-1 Ramsey Road, Shirley, NY 11967, USA, DEIABL547. FOXO1 expression levels were assayed using the rat-specific competitive ELISA kit, MyBioSource, San Diego, CA, MBS749342. The manufacturers’ instructions have been followed in all assay procedures.
Immunohistochemical detection of pancreatic β-arrestin2
An immunohistochemical staining for β-arrestin2 was conducted using the avidin-biotin-peroxidase complex technique. Briefly, 5 µm paraffin-embedded pancreatic tissue sections were deparaffinized, rehydrated, and washed in tap water, followed by blocking of endogenous peroxidase activity using 3% hydrogen peroxide for 10 min. Antigen retrieval was achieved by incubating sections in 10 mmol/L sodium citrate buffer (pH 6.0) in a microwave for 20 min. Sections were then incubated in 2% trypsin at 37°C for 10 min to enhance antigen exposure. Nonspecific binding was blocked using phosphate-buffered saline (PBS) containing 10% normal goat serum.
Sections were incubated with the primary monoclonal antibody against β-arrestin2 (β-arrestin2 (C16D9) Rabbit mAb, Cat. No. #3857, Cell Signaling Technology, Danvers, USA) at a dilution of 1:100 for 30 min at room temperature. After PBS washing, a streptavidin-peroxidase conjugate was applied for 20 min. Visualization of the immune reaction was achieved using 3,3′-diaminobenzidine (DAB) as the chromogen (Dakopatts, Glostrup, Denmark), and sections were counterstained with Harris’ hematoxylin, dehydrated, and cover-slipped.
Both positive and negative controls were included for validation. Rat brain tissue, known for high β-arrestin2 expression,37,38 was used as a positive control and processed in parallel with study samples to verify antibody performance. Negative controls were consistently run by omitting the primary antibody to assess nonspecific staining. The primary antibody used is also validated by the manufacturer for use in formalin-fixed paraffin-embedded tissues.
β-arrestin2 immunoreactivity appeared as brown cytoplasmic staining. The specificity of staining was supported by consistent and expected localization patterns across pancreatic sections and further confirmed by quantitative morphometric analysis. Morphometric analysis of β-arrestin2 immunostaining was performed on sections from three randomly selected rats per group. Using ImageJ v1.51d (NIH & LOCI, University of Wisconsin, USA), the area percentage of DAB-positive staining was quantified at 100× magnification. Brown-stained areas were isolated via binary masking and analyzed relative to a fixed measurement frame.
Histopathological examination
After proper fixation in 10% neutral buffered formalin solution for 48 h, pancreatic tissue specimens were dehydrated in 70%, 80%, 90%, 95%, and 100% ascending grades of ethyl alcohol, cleared in xylol, impregnated, and then embedded in paraffin wax. Five-micron sections were cut using a rotary microtome (Leica RM 2155, England). Sections were stained with either Hematoxylin and Eosin (H&E) or Van Gieson for studying the general histopathological structure of pancreatic tissue and collagen fiber deposition, respectively.
The histopathological lesion grading
Histopathological lesion grading was performed by examining five microscopic fields per H&E-stained pancreatic tissue section from each group, providing a cumulative assessment of pathological alterations. 39 The figure panels represent typical examples and may not capture the full lesion spectrum documented in the scoring table.
Morphometric determination of pancreatic islet area (µm²)
The Pancreatic islet area was morphometrically determined from H&E-stained sections
For each group, six sections were analyzed from three randomly selected rats. All clearly identifiable islets within these sections were photographed under a light microscope at ×400 magnification to enable accurate delineation of islet boundaries. Islet areas were individually measured using ImageJ software (version 1.51d, NIH & LOCI, University of Wisconsin, USA). The mean islet area was then calculated for each group to assess endocrine remodeling. 40
Morphometric determination of collagen fiber area percentage
The area percentage of collagen fibers/µm² was morphometrically assessed in Van Gieson-stained pancreatic sections. For each group, six sections were analyzed from three randomly selected rats. Images were captured at 100× magnification, and collagen fiber-positive areas were quantified using ImageJ software version 1.51d (NIH & LOCI, University of Wisconsin, USA). Fiber-positive (blue-stained) regions were selected, converted to binary masks (red overlay), and measured as a percentage of the total field area using a standardized measurement frame.
Statistical analysis
Statistical analysis was performed using GraphPad Prism version 5.0 (GraphPad Software, Inc., 7825 Fay Avenue, Suite 230, La Jolla, CA 92037 USA). Normality of the data was assessed using the Shapiro–Wilk test and visually inspected via QQ plots. Homoscedasticity was tested with Levene’s test. For data with equal variances, one-way ANOVA followed by Tukey’s post hoc test was used. Welch’s ANOVA was applied in the case of unequal variances. The actual power of the statistical tests was determined using G*Power software, showing power ranging from 98% to 100%. Therefore, any non-significant difference observed was likely attributable to small effect sizes rather than insufficient statistical power or small sample sizes. All results were expressed as mean ± standard error of the mean (SEM). Statistical significance was assumed at p < 0.05 (two-tailed).
Results
Effect on anthropometrical and insulin resistance parameters
As illustrated in Table 1, DXM administration at a dose of 10 mg/kg/day, S.C. for 7 days elicited a profound reduction in BW, BW/tibial length ratio, and BMI, as compared to the control group (p < 0.05). WC didn’t, however, significantly decline. Treatment of DXM-injected rats with either carvedilol (a non-selective β- and α1-blocker), 41 phenylephrine (a selective α1 adrenergic receptor agonist), 42 propranolol (a β-blocker without α1-adrenoceptor blocking actions), 43 or doxazosin (a selective α1-blocker) 44 couldn’t significantly alter almost all body anthropometric parameters. Only phenylephrine could significantly elevate the WC, as compared to the DXM group and the other treatment groups as well (p < 0.05).
Effects of carvedilol (CVD, 10 mg/kg/day, I.P.), phenylephrine (PE, 1 mg/kg/day, I.P.), propranolol (PR, 30 mg/kg/day, I.P.), doxazosin (DZN, 5 mg/kg/day, I.P.), or their respective combinations (PE + CVD, DZN + PR) administered for 7 days on anthropometrical and insulin resistance parameters in dexamethasone (DXM, 10 mg/kg/day, S.C.)-treated rats.

Statistical analysis was performed using one-way ANOVA, followed by Tukey’s post hoc test. Welch’s ANOVA was used in the case of unequal variances. Values are expressed as mean ± SEM. n = 9 for the control and CVD groups. n = 7 for the DXM group. n = 4 for the PE, PE + CVD, PR, and DZN groups. n = 6 for the DZN + PR group. Samples of each group were randomly pooled into three different pools. Statistically significant difference was assumed at p < 0.05 and designated by *control, #DXM, aCVD, bPE, cPE + CVD, dPR, eDZN, and fDZN + PR.
BW: body weight; BMI: body mass index; WC: waist circumference; FSG: fasting serum glucose; FSI: fasting serum insulin; HOMA-IR: Homeostasis Model Assessment of Insulin Resistance.
DXM-induced changes in anthropometrical parameters were associated with a state of IR, as evinced by the significant elevations in FSG and FSI levels and HOMA-IR, as compared to the control group (p < 0.05). Moreover, DXM-treated rats showed a compensatory significant expansion in pancreatic islet area, as compared to the control group (p < 0.05). All treatment groups unpredictably displayed non-significant alterations in FSG levels, as compared to the DXM group (p < 0.05). Nevertheless, there seemed to be a trend for either carvedilol, phenylephrine/carvedilol, propranolol, or doxazosin to lower the FSG levels versus the DXM group. Doxazosin elicited the most pronounced reducing effect on FSG, and such effect was significant as compared to that of either phenylephrine or doxazosin/propranolol combination, which both rather elevated the FSG level to an extent significantly surpassing that of the control group (p < 0.05). On the other hand, FSI levels were significantly reduced in all treatment groups versus the DXM group (p < 0.05). Apart from phenylephrine, HOMA-IR was significantly reduced with all other treatments, as compared to the DXM group (p < 0.05).
Indeed, doxazosin and its combination with propranolol prompted the most outstanding effects, with the combination group far more approaching the near normal insulin levels and both groups restoring the normal insulin sensitivity of the control rats. The effects of propranolol and carvedilol didn’t differ significantly. Interestingly, the insulin-lowering effect of carvedilol was significantly reduced upon preinjection of phenylephrine, and that of propranolol was enhanced upon co-administration with doxazosin, yet with an accompanying non-significant alteration in insulin sensitivity.
Effect on apoptosis markers
As depicted in Figure 2, significant downregulation of Bcl-2 expression and upregulation of both BAX expression and BAX/Bcl-2 expression ratio in the whole pancreas were demonstrated in DXM-treated rats versus the control group (p < 0.05), pointing to a state of enhanced apoptosis and reduced cell survival. DXM-induced Bcl-2 downregulation could only be significantly reversed by either propranolol, doxazosin, or their combined administration. Yet, BAX and BAX/Bcl-2 expression ratio were significantly downregulated in all treatment groups, as compared to the DXM group (p < 0.05). As aforementioned, doxazosin and its combination with propranolol approached the normal healthy state seen in the control group, with their impact on BAX, Bcl-2, and BAX/Bcl-2 expression ratio being significant, as compared to either carvedilol, phenylephrine, or both and superior to that of propranolol. However, differences in the measured parameters between propranolol and carvedilol-treated rats have still been non-significant. Seemingly, the effects of carvedilol were only a bit weakened upon preinjection of phenylephrine, pointing to a minor role of α1-adrenoceptor blockade in mediating the anti-apoptotic effects of carvedilol. In the same regard, co-administration of doxazosin with propranolol remarkably potentiated the effects of propranolol.

Changes in pancreatic apoptotic markers. Graphical presentation of the B-cell lymphoma-2-Associated X Protein (BAX) expression (a), B-cell lymphoma-2 (Bcl-2) expression (b), and BAX/Bcl-2 expression ratio (c). Control: Rats intraperitoneally administered both distilled water and vehicle (DMSO: tween 80: water in a volume ratio of 1:1:8) for 7 days. DXM: Rats administered DXM dissolved in distilled water (10 mg/kg/day, S.C.) and intraperitoneally received the vehicle for 7 days. CVD: Rats received carvedilol dissolved in the vehicle (10 mg/kg/day, I.P.), intraperitoneally administered distilled water, and treated with DXM 2 h later for 7 days. PE: Rats administered phenylephrine dissolved in distilled water (1 mg/kg/day, I.P.), intraperitoneally received the vehicle, and treated with DXM 2 h later for 7 days. PE + CVD: Rats given phenylephrine followed by carvedilol 30 min later and treated with DXM 2 h later for 7 days. PR: Rats treated with propranolol dissolved in the vehicle (30 mg/kg/day, I.P.), intraperitoneally administered distilled water, and treated with DXM 2 h later for 7 days. DZN: Rats received doxazosin dissolved in the vehicle (5 mg/kg/day, I.P.), intraperitoneally administered distilled water, and treated with DXM 2 h later for 7 days. DZN + PR: Rats given doxazosin followed by propranolol 30 min later, intraperitoneally administered distilled water, and treated with DXM 2 h later for 7 days. Statistical analysis was performed using one-way ANOVA, followed by Tukey’s post hoc test. Welch’s ANOVA was used in the case of unequal variances. Values are expressed as mean ± SEM. n = 9 for the control and CVD groups. n = 7 for the DXM group. n = 4 for the PE, PE + CVD, PR, and DZN groups. n = 6 for the DZN + PR group. Samples of each group were randomly pooled into three different pools. Statistically significant difference was assumed at p < 0.05 and designated by *control, #DXM, aCVD, bPE, cPE + CVD, dPR, eDZN, and fDZN + PR.
Effect on collagen fiber deposition
Administration of DXM resulted in a significant increase in area% of collagen fibers/µm2 in exocrine pancreas, as compared to the control group (p < 0.05). Curiously, the % fibrotic area in pancreatic islets was, rather, markedly reduced in DXM-treated rats versus the control group (p < 0.05). There were non-significant differences in the % fibrotic area in pancreatic islets among the treatment groups. Apart from carvedilol, all other treatment groups could markedly reduce the % fibrotic area in the exocrine pancreas versus the DXM group (p < 0.05; Figure 3).

Changes in pancreatic fibrotic response. Representative photomicrographs of collagen deposits in the Van Gieson-stained sections of pancreatic tissue (× 100) (a). Graphical presentation of the area % of collagen fibers/μm2 in pancreatic islets (n = 3) (b) and in exocrine pancreas (n = 6) (c). Control: Rats intraperitoneally administered both distilled water and vehicle (DMSO: tween 80: water in a volume ratio of 1:1:8) for 7 days. DXM: Rats administered DXM dissolved in distilled water (10 mg/kg/day, S.C.) and intraperitoneally received the vehicle for 7 days. CVD: Rats received carvedilol dissolved in the vehicle (10 mg/kg/day, I.P.), intraperitoneally administered distilled water, and treated with DXM 2 h later for 7 days. PE: Rats administered phenylephrine dissolved in distilled water (1 mg/kg/day, I.P.), intraperitoneally received the vehicle, and treated with DXM 2 h later for 7 days. PE + CVD: Rats given phenylephrine followed by carvedilol 30 min later and treated with DXM 2 h later for 7 days. PR: Rats treated with propranolol dissolved in the vehicle (30 mg/kg/day, I.P.), intraperitoneally administered distilled water, and treated with DXM 2 h later for 7 days. DZN: Rats received doxazosin dissolved in the vehicle (5 mg/kg/day, I.P.), intraperitoneally administered distilled water, and treated with DXM 2 h later for 7 days. DZN + PR: Rats given doxazosin followed by propranolol 30 min later, intraperitoneally administered distilled water, and treated with DXM 2 h later for 7 days. Statistical analysis was performed using one-way ANOVA, followed by Tukey’s post hoc test. Welch’s ANOVA was used in the case of unequal variances. Values are expressed as mean ± SEM. Statistically significant difference was assumed at p < 0.05 and designated by *control, #DXM, aCVD, bPE, cPE + CVD, dPR, eDZN, fDZN + PR.
Effect on cAMP, PKA-ERK1/2-CREB, and PKB-FOXO1 signaling pathways
As represented in Figure 4, levels of cAMP in the entire pancreatic tissues from DXM-treated rats were significantly decreased, a matter which was unpredictably associated with upregulated PKA, ERK1/2, and CREB expression levels (p < 0.05). It was in the same context that DXM administration resulted in significantly higher PKB activity and lower FOXO1 expression levels versus the control group (p < 0.05).

Changes in cyclic adenosine monophosphate (cAMP), protein kinase A (PKA), extracellular signal-regulated kinases 1 and 2 (ERK1/2), and cAMP-responsive element-binding protein (CREB) expression levels, protein kinase B (PKB) activity, and forkhead box O1 (FOXO1) expression in pancreatic tissue. Graphical presentation of cAMP (a), PKA (b), ERK1/2 (c), and CREB (d) expression levels, PKB activity (e), and FOXO1 expression (f). Control: Rats intraperitoneally administered both distilled water and vehicle (DMSO: tween 80: water in a volume ratio of 1:1:8) for 7 days. DXM: Rats administered DXM dissolved in distilled water (10 mg/kg/day, S.C.) and intraperitoneally received the vehicle for 7 days. CVD: Rats received carvedilol dissolved in the vehicle (10 mg/kg/day, I.P.), intraperitoneally administered distilled water, and treated with DXM 2 h later for 7 days. PE: Rats administered phenylephrine dissolved in distilled water (1 mg/kg/day, I.P.), intraperitoneally received the vehicle, and treated with DXM 2 h later for 7 days. PE + CVD: Rats given phenylephrine followed by carvedilol 30 min later and treated with DXM 2 h later for 7 days. PR: Rats treated with propranolol dissolved in the vehicle (30 mg/kg/day, I.P.), intraperitoneally administered distilled water, and treated with DXM 2 h later for 7 days. DZN: Rats received doxazosin dissolved in the vehicle (5 mg/kg/day, I.P.), intraperitoneally administered distilled water, and treated with DXM 2 h later for 7 days. DZN + PR: Rats given doxazosin followed by propranolol 30 min later, intraperitoneally administered distilled water, and treated with DXM 2 h later for 7 days. Statistical analysis was performed using one-way ANOVA, followed by Tukey’s post hoc test. Welch’s ANOVA was used in the case of unequal variances. Values are expressed as mean ± SEM. n = 9 for the control and CVD groups. n = 7 for the DXM group. n = 4 for the PE, PE + CVD, PR, and DZN groups. n = 6 for the DZN + PR group. Samples of each group were randomly pooled into three different pools. Statistically significant difference was assumed at p < 0.05 and designated by *control, #DXM, aCVD, bPE, cPE + CVD, dPR, eDZN, and fDZN + PR.
The current study has revealed that only propranolol, doxazosin, and their combination could significantly restore the cAMP expression levels versus the DXM group. Nevertheless, PKA, ERK1/2, and CREB expression levels were significantly reduced in all treatment groups, as compared to the DXM group (p < 0.05). Although PKB activity was significantly reduced with all given treatments, FOXO1 expression was only significantly restored by either propranolol, doxazosin, or their dual administration and merely slightly elevated with carvedilol, phenylephrine, and their combination, as compared to the DXM group (p < 0.05).
Importantly, the most pronounced effects were indeed elicited by both doxazosin and the doxazosin/propranolol combination, bringing the values back close to the normal levels seen in the control group. Such effects surpassed those of propranolol and were apparently superior to those of carvedilol, phenylephrine, or their combination. Non-significant differences in the measured parameters were, however, observed between propranolol and carvedilol-treated rats. It’s noteworthy that administration of phenylephrine before carvedilol slightly attenuated its effects, further corroborating the minor role of α1-adrenoceptor blockade in the carvedilol-mediated regulation of cell signaling pathways in pancreatic tissue. It was in the same context to note a significant potentiating effect upon co-administration of doxazosin with propranolol, as compared to that of propranolol administered alone.
Effect on pancreatic immunoexpression of β-arrestin2
Immunohistochemical detection of β-arrestin2 in pancreatic tissue was performed. As depicted in Figure 5, the % level of β-arrestin2 didn’t considerably differ between endocrine and exocrine pancreatic tissues from control rats. There, however, existed a differential expression of β-arrestin2 in pancreatic islets versus exocrine pancreas from DXM-treated rats. % Level of β-arrestin2 was remarkably reduced in pancreatic islets and was, however, surprisingly elevated in exocrine pancreas from DXM-treated rats, as compared to the control group (p < 0.05). Unexpectedly, the % level of β-arrestin2 in pancreatic islets from DXM-treated rats was even more markedly reduced with all given treatments except for carvedilol. The latter significantly restored β-arrestin2 expression in pancreatic islets, as compared to the DXM group (p < 0.05). Apart from carvedilol, the % level of β-arrestin2 in exocrine pancreas from DXM-treated rats was significantly reduced with all other treatments, as compared to the DXM group (p < 0.05).
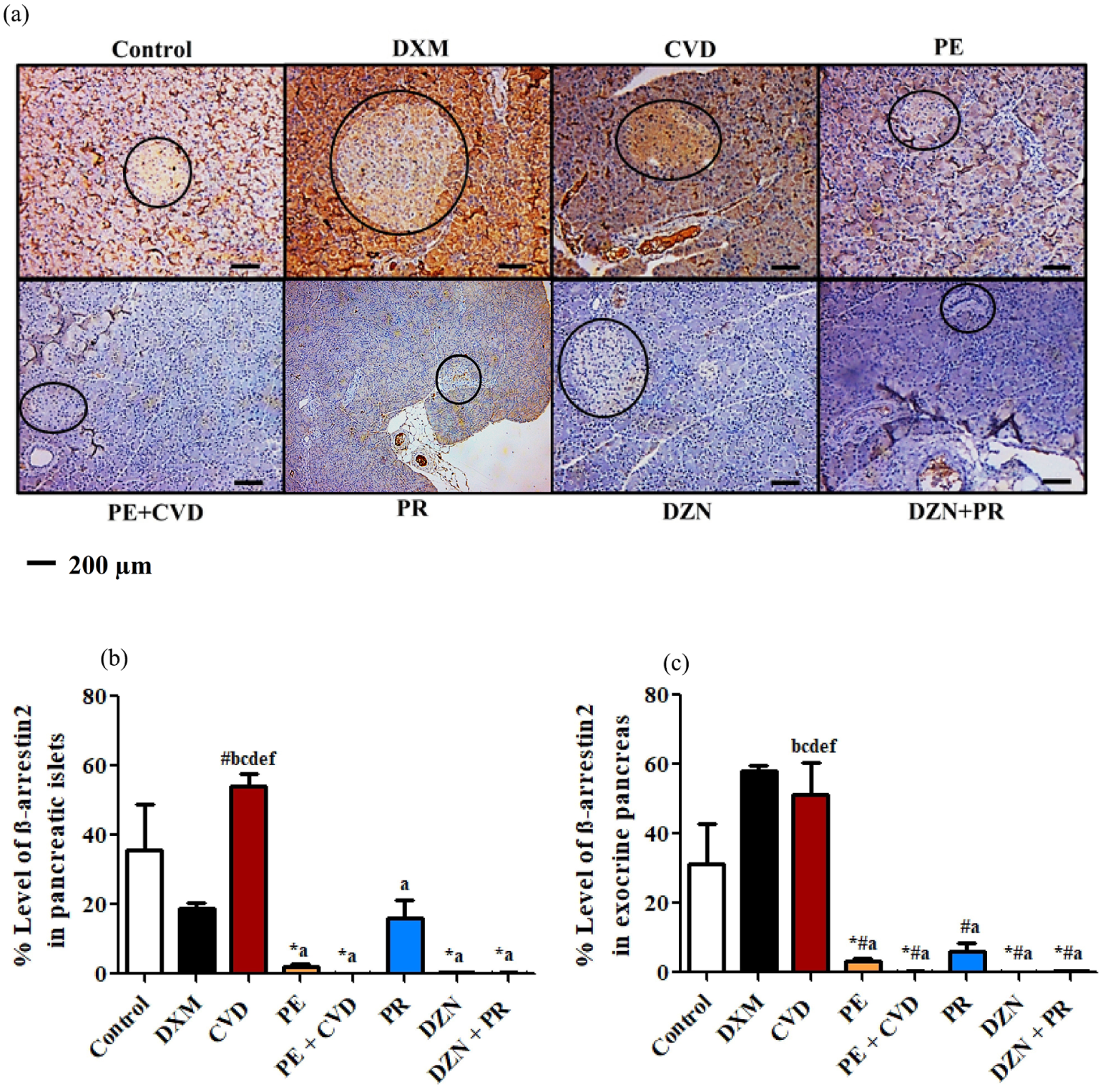
Changes in the % level of β-arrestin2. Representative photomicrographs of immuno-stained sections of pancreatic tissue (×100) (a). Graphical presentation of % Level of β-arrestin2 in pancreatic islets (b) and in exocrine pancreas (c). Control: Rats intraperitoneally administered both distilled water and vehicle (DMSO: tween 80: water in a volume ratio of 1:1:8) for 7 days. DXM: Rats administered DXM dissolved in distilled water (10 mg/kg/day, S.C.) and intraperitoneally received the vehicle for 7 days. CVD: Rats received carvedilol dissolved in the vehicle (10 mg/kg/day, I.P.), intraperitoneally administered distilled water, and treated with DXM 2 h later for 7 days. PE: Rats administered phenylephrine dissolved in distilled water (1 mg/kg/day, I.P.), intraperitoneally received the vehicle, and treated with DXM 2 h later for 7 days. PE + CVD: Rats given phenylephrine followed by carvedilol 30 min later and treated with DXM 2 h later for 7 days. PR: Rats treated with propranolol dissolved in the vehicle (30 mg/kg/day, I.P.), intraperitoneally administered distilled water, and treated with DXM 2 h later for 7 days. DZN: Rats received doxazosin dissolved in the vehicle (5 mg/kg/day, I.P.), intraperitoneally administered distilled water, and treated with DXM 2 h later for 7 days. DZN + PR: Rats given doxazosin followed by propranolol 30 min later, intraperitoneally administered distilled water, and treated with DXM 2 h later for 7 days. Statistical analysis was performed using one-way ANOVA, followed by Tukey’s post hoc test. Welch’s ANOVA was used in the case of unequal variances. Values are expressed as mean ± SEM (n = 3). Statistically significant difference was assumed at p < 0.05 and designated by *control, #DXM, aCVD, bPE, cPE + CVD, dPR, eDZN, and fDZN + PR.
Correlations
As depicted in Figure 6, a significant inverse correlation was found between cAMP expression level in pancreatic tissue and % level of β-arrestin2 in exocrine pancreas (Pearson r = −0.44). PKA, ERK1/2, and CREB expression levels were, however, significantly and directly correlated to the % level of β-arrestin2 (Pearson r = 0.60, 0.52, and 0.51, respectively). Likewise, there existed a significant direct correlation between PKB activity and the % level of β-arrestin2 (Pearson r = 0.50). FOXO1 expression level was not significantly, yet inversely correlated to the % level of β-arrestin2 (Pearson r = −0.38). The area % of collagen fibers/μm2 in either pancreatic islets or exocrine pancreas was significantly and directly correlated to the % level of β-arrestin2 in the respective tissues (Pearson r = 0.46 and 0.65, respectively).

Correlation of cyclic adenosine monophosphate (cAMP) expression (a), protein kinase A (PKA) expression (b), extracellular signal-regulated kinases 1 and 2 (ERK1/2) expression (c), cAMP-responsive element-binding protein (CREB) expression (d), protein kinase B (PKB) activity (e), and forkhead box O1 (FOXO1) expression (f) with % level of β-arrestin2 in exocrine pancreas and correlation of area % of collagen fibers/μm2 with % level of β-arrestin2 in pancreatic islets (g) and in exocrine pancreas (h) (p < 0.05).
Effect on the histopathological changes of pancreatic tissue
Blinded qualitative assessment of H&E-stained pancreatic sections (Figure 7), along with semi-quantitative lesion scoring (Table 2), revealed that the control group exhibited normal islets of Langerhans and pancreatic acini, with no significant degenerative changes. However, mild interstitial edema and inflammatory exudate were observed in a few sections, which are commonly seen in control animals and may reflect nonspecific background inflammation, possibly resulting from routine handling or environmental factors. These changes were not considered indicative of any pathological condition. The DXM group displayed the most severe pathological alterations, including mild inflammatory infiltration and moderate degenerative/necrotic changes in the islets with remnants of nuclear elements, mild degeneration of pancreatic acini, moderate interstitial edema with inflammatory exudate, and moderate vascular congestion—indicative of a pronounced glucocorticoid-induced pancreatic insult.
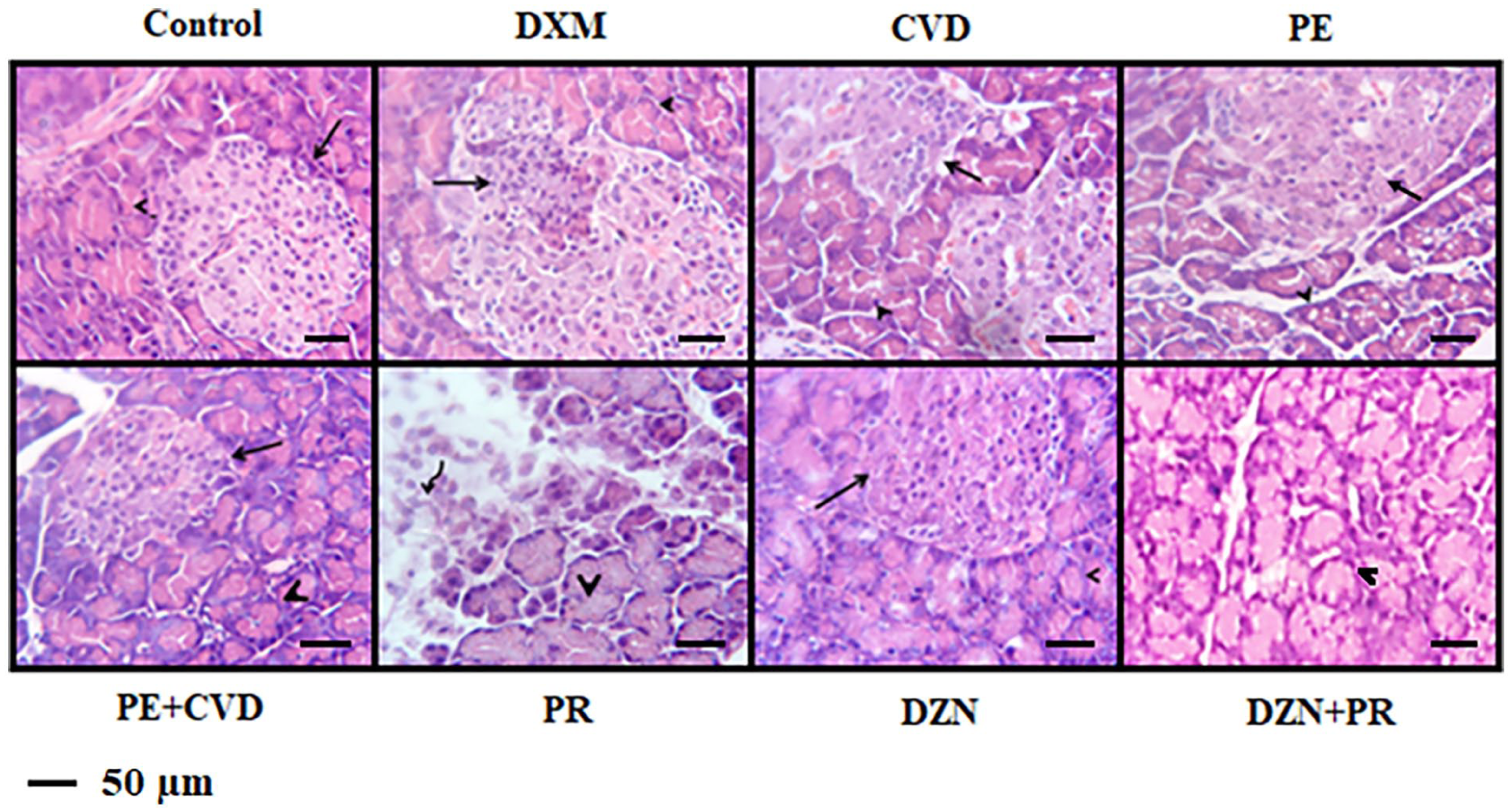
Changes in the general histopathological structure of pancreatic tissue specimens. Representative light photomicrographs of H&E-stained pancreatic sections (×400) are depicted from:
Semi-quantitative scoring of histopathological pancreatic lesions in dexamethasone (DXM, 10 mg/kg/day, S.C.)-treated rats following 7-day treatment with carvedilol (CVD, 10 mg/kg/day, I.P.), phenylephrine (PE, 1 mg/kg/day, I.P.), propranolol (PR, 30 mg/kg/day, I.P.), doxazosin (DZN, 5 mg/kg/day, I.P.), or their respective combinations (PE + CVD, DZN + PR).

Histopathological lesion grading was performed by examining five microscopic fields per H&E-stained tissue section from each group, providing a cumulative assessment of pathological alterations. Scoring was defined as follows: (−) no lesion, (+) mild, (++) moderate, (+++) severe tissue alterations.
The carvedilol group retained moderate inflammatory infiltration in the islets without evidence of degeneration or necrosis, whereas the phenylephrine group showed mild islet degenerative and necrotic changes. The exocrine pancreas of both groups exhibited moderate acinar degeneration with mild necrosis, evidenced by cytoplasmic vacuolations and pyknotic nuclei in affected acinar cells. No notable interstitial edema, inflammatory exudate, or vascular congestion was observed in either group. The phenylephrine/carvedilol combination and doxazosin groups exhibited preserved pancreatic architecture, with no notable pathological lesions in either the endocrine or exocrine compartments, nor in the surrounding stroma or vasculature. The propranolol group demonstrated moderate acinar degeneration with mild necrosis, accompanied by dissociation of pancreatic acini and periacinar lymphoplasmacytic infiltrations, indicative of tissue injury. Marked interstitial edema with inflammatory exudate was observed, particularly evident around the pancreatic lobules. Mild vascular congestion was also noted. The doxazosin/propranolol combination group revealed moderate inflammatory infiltration and mild degeneration in the islets, mild necrosis in both islets and acini, and mild vascular congestion, with otherwise preserved histoarchitecture.
Discussion
Glucocorticoids are renowned for their ubiquitous use in various clinical settings and the abundant adverse effects they may exert. 45 In the present study, subcutaneous administration of DXM (10 mg/kg/day) for 7 days significantly reduced BW, BW/tibial length ratio, and BMI, likely mediated by increased plasma leptin and restricted food intake. 46 These anthropometric changes were accompanied by clear signs of IR, as reflected by significant increases in FSG, FSI, and HOMA-IR indices, alongside compensatory enlargement of pancreatic islet area relative to controls.
These findings align with previous reports that DXM promotes IR and induces compensatory β-cell proliferation and hyperinsulinemia, ultimately leading to β-cell dysfunction, glucose homeostasis disruption, and hyperglycemia.4,46,47 In addition to metabolic disturbances, DXM is known to exert direct cytotoxic effects on pancreatic tissue, including necrosis and pancreatitis.48,49 Biochemically, DXM upregulates pro-apoptotic BAX and downregulates anti-apoptotic Bcl-2, promoting apoptosis in pancreatic acinar cells. 50
Consistently, our histopathological findings revealed widespread pancreatic damage in the DXM group, including inflammatory cell infiltration, islet degeneration and necrosis, acinar injury, interstitial edema, and vascular congestion. These alterations coincided with a significant increase in the pancreatic Bax/Bcl-2 expression ratio, supporting enhanced apoptotic activity. Furthermore, the associated fibrosis likely reflects activation of pancreatic stellate cells, which differentiate into collagen-producing myofibroblasts in response to apoptotic signaling. 51 Although not explicitly measured in the current study, previous reports indicate that stellate cell-derived transforming growth factor-β may promote exocrine cell remodeling while suppressing endocrine cell differentiation, 52 offering a plausible explanation for the observed increase in collagen deposition in the exocrine pancreas and its relative reduction in the islets.
Unraveling the mechanistic basis of these DXM-induced effects, we investigated β-arrestin2 crosstalk with key intracellular signaling pathways, including cAMP, PKA-ERK1/2-CREB, and PKB-FOXO1. Intracellular cAMP signaling is known to exert anti-fibrotic effects, 53 whereas activation of ERK1/2 and its downstream effector CREB has been implicated in promoting fibrotic and proliferative responses. 54 In line with the enhanced fibrotic remodeling observed in the exocrine pancreas of DXM-treated rats, we found a significant reduction in pancreatic cAMP levels, alongside increased expression of PKA, ERK1/2, and CREB.
Hyperinsulinemia has also been shown to activate PKB and inhibit FOXO1 in cultured pancreatic stellate cells, thereby promoting cell proliferation and fibrosis. 55 Accordingly, the elevated PKB activity and reduced FOXO1 expression observed in our study may further explain the marked expansion of pancreatic islet area 56 and heightened fibrotic responses in exocrine regions. 55
Together, these findings support the concept that although glucocorticoids sensitize cells to apoptosis, 45 they also simultaneously trigger proliferative and fibrogenic signaling pathways.54,57,58 Regarding the role of β-arrestin2 in this context, the current study demonstrates, for the first time, a potential role for β-arrestin2 in driving pancreatic fibrotic remodeling. Specifically, the area percentage of collagen fibers/μm² in both islets and exocrine regions was significantly and directly correlated with the respective β-arrestin2 expression levels—both of which were reduced in islets and elevated in the exocrine pancreas of DXM-treated rats.
We acknowledge that the divergent expression patterns of β-arrestin2—its reduction in pancreatic islets and elevation in the exocrine pancreas—may initially appear contradictory. However, this likely reflects a novel and potentially meaningful tissue-specific redistribution of β-arrestin2 in response to DXM-induced metabolic stress. A possible explanation is that endocrine cells are more sensitive to glucocorticoids than exocrine cells because they express significantly more glucocorticoid receptor mRNA transcripts. 59 This spatial distinction may therefore suggest differential regulatory roles of β-arrestin2 in endocrine versus exocrine compartments, possibly shaped by distinct receptor distributions, cellular stress responses, or local microenvironmental cues. It is worth illuminating, here, that β-arrestins are critical signaling scaffolds that play central roles in regulating glucose homeostasis, including the maintenance of euglycemia and peripheral insulin sensitivity. 60 In this context, the marked reduction in β-arrestin2 expression within pancreatic islets appears to be mechanistically linked to the previously reported state of IR with hyperglycemia and hyperinsulinemia. Conversely, the upregulated β-arrestin2 expression in the exocrine pancreas may contribute to the enhanced fibrotic response, as supported by the significant correlation between β-arrestin2 levels and fibrotic area percentage. These compartment-specific expression patterns underscore the dual and spatially distinct roles of β-arrestin2 in modulating both endocrine dysfunction and fibrotic remodeling in the pancreas under glucocorticoid-induced metabolic stress. While the underlying mechanisms remain to be fully elucidated, this observation opens new avenues for future research investigating the compartment-specific roles of β-arrestin2 in pancreatic pathophysiology.
The pancreas is composed predominantly of exocrine tissue (approximately 95%), with endocrine tissue making up less than 5%. 61 Therefore, islets contributed minimally to the whole pancreatic protein expression levels of either cAMP, PKA, ERK1/2, CREB, or FOXO1, and even PKB activity. The DXM-induced modulations of these pathways were significantly correlated with the upregulated β-arrestin2 expression in the exocrine pancreas. This aligns with prior evidence indicating that β-arrestin2 reduces intracellular cAMP levels, 62 activates the ERK-CREB axis in a PKA-independent manner, 63 and contributes to PKB activation, 20 while also suppressing FOXO1 expression via PKB-independent mechanisms. 64 Given the established roles of these pathways in fibrogenesis, our findings provide a mechanistic basis for β-arrestin-mediated fibrotic responses. It is worth emphasizing that the mechanistic associations drawn between β-arrestin2 and the examined apoptotic and fibrotic signaling pathways pertain specifically to the exocrine pancreas, and are mechanistically distinct from its role in endocrine-mediated regulation of glucose homeostasis.
Nonetheless, we acknowledge that β-arrestin2 operates within a broader signaling milieu and that key pathways such as transforming growth factor-β and inflammatory cytokines likely contribute significantly to the apoptotic and fibrotic landscape. 65 Further studies are warranted to explore how these parallel and intersecting signals integrate with β-arrestin2-mediated mechanisms. Such investigations could involve cytokine profiling, the use of targeted pathway inhibitors, or genetic manipulation tools to more comprehensively elucidate the molecular underpinnings of pancreatic fibrosis in this context. In fact, we used β-arrestin2 expression as a proxy for biased β-arrestin2 signaling, which we believe is a biologically meaningful starting point for distinguishing drug effects on downstream fibrotic outcomes.
To explore the involvement of β- and α1-adrenoceptors in pancreatic regulation of glucose homeostasis and cell survival under insulin-resistant conditions, we evaluated the effects of carvedilol, phenylephrine, phenylephrine + carvedilol, propranolol, doxazosin, and doxazosin + propranolol in DXM-treated rats. Carvedilol is a third-generation non-selective β- and α1-blocker. 41 Phenylephrine is a selective α1-adrenoceptor agonist. 42 Propranolol is a β-blocker without α1-adrenoceptor blocking actions. 43 Doxazosin is a selective α1-blocker. 44
Regarding glucose metabolism, β-blockers are known to impair β2-adrenoceptor-mediated insulin release. 66 By suppressing β2-stimulated hepatic glycogenolysis, gluconeogenesis, and glucagon secretion, they may promote decreased blood glucose levels and improved insulin sensitivity. 67 In this study, the reducing effect of carvedilol and propranolol on FSG, FSI, and HOMA-IR didn’t significantly differ, suggesting the role of β-adrenoceptor blockade in mediating their effects. Notably, phenylephrine preinjection attenuated the effects of carvedilol, suggesting that α1-adrenoceptor blockade contributes meaningfully to carvedilol’s metabolic benefits. Phenylephrine alone slightly elevated FSG and significantly reduced FSI. This observation aligns with its dual effects. Hepatic α1-adrenoceptor stimulation promotes hyperglycemia,68,69 while α1-activation in pancreatic islets inhibits insulin secretion.70,71 These effects together suggest a modest net improvement in insulin sensitivity. Doxazosin has previously been shown to lower both glucose and insulin levels in hypertensive patients 72 and to improve IR. 73 Consistent with this, either doxazosin or propranolol in our model reduced the FSG, which was rather elevated by their co-administration, owing to the severely reduced FSI. Nevertheless, FSG was only slightly elevated versus the DXM group, explaining the marked improvement in insulin sensitivity elicited by the combined administration.
The current study is the first to report an anti-apoptotic effect of either agent in pancreatic tissues from DXM-treated rats, downregulating both BAX and BAX/Bcl-2 expression ratio. These findings align with previous research in other models. For instance, phosphatidic acid-induced Bcl-2 expression in HeLa cells was further enhanced by pretreatment with propranolol, 74 and carvedilol was shown to inhibit ethylene glycol-induced upregulation of BAX while preserving Bcl-2 expression in rat kidneys. 75 Likewise, phenylephrine abrogated hypoxia and serum deprivation-induced upregulation of the BAX: Bcl-2 expression ratio in neonatal cardiomyocytes. 76 However, contrasting effects have been reported for doxazosin, which induced apoptosis in cultured cardiomyocytes independently of α1-adrenoceptor blockade. 77 Apparently, the same drug may exert contrasting effects on apoptosis, depending on its concentration and cell type. 78
Regarding fibrotic changes, no significant differences were observed in islet collagen deposition among the treatment groups. However, all treatments except carvedilol significantly reduced the fibrotic area in the exocrine pancreas compared to the DXM group. This aligns with prior findings demonstrating the antifibrotic effects of propranolol in a murine model of nonsinusoidal liver fibrosis. 79 Although previous studies reported antifibrotic actions of carvedilol in hepatic and myocardial tissues,80–82 such effects were not evident in our pancreatic model. The divergent outcomes may reflect tissue-specific signaling dynamics or differential β-arrestin2 activity. Notably, high-dose phenylephrine (25 mg/kg/day) has been associated with cardiac fibrosis in rats, 83 whereas our study used a much lower dose (1 mg/kg/day), potentially explaining the absence of profibrotic effects. In agreement with our results, doxazosin has consistently shown antifibrotic activity in various tissues.84,85
Mechanistically, this study is the first to report that carvedilol, phenylephrine, propranolol, and doxazosin reversed the DXM-induced alterations in key pancreatic signaling pathways. Specifically, all four agents significantly upregulated cAMP while downregulating PKA, ERK1/2, and CREB expression, along with reduced PKB activity and increased FOXO1 expression. These coordinated modulations likely underlie the reversal of islet hypertrophy and the antifibrotic effects observed with all treatments—except carvedilol.55,56,86–88 The observed reductions in PKA, ERK1/2, and CREB with β-blockers align with prior studies demonstrating that β-adrenoceptor activation promotes PKA-ERK1/2-CREB signaling and induces cell proliferation via CREB activation, 89 as well as PKB phosphorylation. 90 This supports our finding that carvedilol and propranolol attenuate these pro-growth and survival pathways.
Interestingly, FOXO1 levels have paradoxically been reported to increase upon β-adrenoceptor stimulation in vitro, 91 suggesting tissue- and context-dependent effects. α1-Adrenoceptor activation has similarly been associated with ERK1/2 activation and PKB phosphorylation,92,93 which may explain the downregulation of these signaling mediators following doxazosin treatment. However, the phenylephrine-induced changes may not be entirely attributable to α1-adrenoceptor stimulation. In cardiomyocytes, phenylephrine (50 μM) failed to induce PKB phosphorylation, caused FOXO1 dephosphorylation, and upregulated FOXO1 with chronic exposure, 94 suggesting an alternate, possibly receptor-independent regulatory mechanism.
Concerning β-arrestin2 signaling, the downregulation of β-arrestin2 expression observed with phenylephrine, propranolol, and doxazosin may have contributed to their anti-fibrotic effects. In contrast, the carvedilol-induced upregulation of β-arrestin2 may underlie its failure to counteract the DXM-induced pancreatic fibrosis. Although direct demonstration of β-arrestin2-biased signaling was beyond the scope of this pharmacopathological study, our findings are consistent with the well-established β-arrestin2-biased signaling property of carvedilol, as previously reported in the literature. 19 While both carvedilol and propranolol act as β-adrenoceptor blockers, carvedilol uniquely possesses α1-adrenoceptor blockade and β-arrestin2 signaling bias, unlike propranolol, which lacks such bias. 41 The divergence in β-arrestin2 expression and corresponding modulation of fibrotic responses likely reflects these pharmacodynamic distinctions. Supporting this interpretation, carvedilol has been shown to upregulate β-arrestin2 in neural tissues, 95 while propranolol has been reported to antagonize β-arrestin2-mediated effects, as demonstrated in a recent study where β-arrestin2 overexpression in the infralimbic prefrontal cortex enhanced extinction of cocaine reward memory—an effect abolished by propranolol. 96 Additionally, doxazosin and phenylephrine have been reported to downregulate β-arrestin2 expression in the cerebral cortex and cultured cells, respectively. 97
Collectively, our findings suggest a potentially context-dependent, pro-fibrotic role of β-arrestin2 signaling in the exocrine pancreas. The observed variations in fibrosis across treatment groups underscore the importance of tissue-specific β-arrestin2 dynamics in guiding therapeutic potential and warrant further mechanistic studies to clarify its precise regulatory role in pancreatic remodeling.
In general, the comparable effects of carvedilol and propranolol highlight the central role of β-adrenoceptor blockade in mediating their pharmacological actions. The modest attenuation of carvedilol’s effects following phenylephrine pretreatment suggests only a minor contribution from its α1-adrenoceptor blocking activity. This observation is consistent with the reported finding that carvedilol is approximately 10 times less potent at α1-adrenoceptors than at β-adrenoceptors, 98 which may in part explain its significantly weaker effects compared to doxazosin in our model, despite its dual blocking properties. Interestingly, while the most pronounced improvements induced by the doxazosin/propranolol combination were comparable to those of doxazosin alone, co-administration significantly enhanced the effects of propranolol, suggesting a potential synergistic benefit of combining β- and α1-blockade.
It is worth emphasizing that the superior protective effects of doxazosin relative to either carvedilol or propranolol may stem not only from its selective and more potent α1-adrenoceptor antagonism but also from its lack of β-arrestin2 signaling bias. This may have allowed for more favorable modulation of downstream fibrotic pathways, which are implicated in tissue remodeling under glucocorticoid stress. Furthermore, an important vascular perspective should be incorporated. While direct evidence linking doxazosin or α1-adrenoceptor blockade to increased pancreatic blood flow is limited, several studies provide indirect support for this hypothesis. Doxazosin has been reported to improve insulin sensitivity in diabetic hypertensive patients, potentially via enhanced peripheral perfusion. 99 Prazosin, an α1-blocker, has been shown to reduce portal pressure and hepatic vascular resistance in cirrhotic patients, leading to increased hepatic blood flow. Given the anatomical and physiological similarities within the splanchnic circulation, it’s plausible that α1-blockade could similarly enhance pancreatic perfusion. 100
On the other hand, propranolol has actually been reported to significantly reduce the blood flow to the stomach, colon, pancreas, and mesentery, but not to the small intestine and spleen. This was the result of a reduced cardiac output and of splanchnic vasoconstriction, evidenced by a marked increase in splanchnic arteriolar resistance. 101 Together, these findings emphasize that receptor subtype selectivity, drug potency, β-arrestin2 signaling bias, and impact on organ perfusion are critical determinants of therapeutic outcomes and provide mechanistic insight into the observed superiority of doxazosin in the context of glucocorticoid-induced pancreatic pathology.
Ultimately, histopathological assessment revealed varying degrees of protection across treatment groups. Carvedilol and phenylephrine monotherapies yielded only partial histological recovery in both islet and acinar regions. In contrast, the phenylephrine/carvedilol combination and doxazosin monotherapy preserved overall pancreatic architecture, with no significant lesions in either the endocrine or exocrine compartments. The propranolol group demonstrated prominent acinar injury, which was modestly improved by adding doxazosin. Although the doxazosin/propranolol combination achieved the most pronounced improvements in molecular signaling parameters—such as the BAX/Bcl-2 ratio, cAMP, PKA-ERK1/2-CREB, and PKB-FOXO1 pathways—its histopathological recovery was slightly less robust than that observed with doxazosin alone. This discrepancy could reflect a temporal disconnect between early signaling recovery and complete tissue remodeling. Indeed, the consistent superiority of doxazosin across molecular and structural endpoints affirms the dominant protective role of selective α1-blockade in this model. Hence, the apparent paradox of enhanced histological outcomes with phenylephrine/carvedilol over carvedilol alone, despite the phenylephrine-induced attenuation of carvedilol’s α1-blockade, could reflect a complex interaction between α1-adrenoceptor stimulation and β-arrestin2 signaling dynamics. Overall, the findings underscore the context-dependent role of β-arrestin2 and the impact of receptor selectivity and signaling bias on both molecular and structural outcomes during glucocorticoid-induced pancreatic injury.
In conclusion, our findings offer several novel insights that advance the understanding of adrenoceptor signaling in pancreatic regulation under glucocorticoid-induced metabolic stress. DXM administration resulted in glucose homeostasis disruption, compensatory pancreatic islet mass expansion, inflammatory infiltration, islet degeneration/necrosis, acinar damage, interstitial edema, and vascular congestion, as well as pronounced pancreatic apoptosis and fibrosis. For the first time, we identify a compartment-specific alteration in β-arrestin2 expression—markedly reduced in islets and elevated in the exocrine pancreas—suggesting a differential compartmental role of β-arrestin2 in islet versus non-islet regions. This spatial divergence may provide a basis for future studies aiming to explore the selective vulnerability or adaptive responses of pancreatic islet cells under metabolic stress conditions.
Mechanistically, we delineate a distinct signaling axis involving cAMP, PKA-ERK1/2-CREB, and PKB-FOXO1. While individual components of this pathway have been studied, their integrated orchestration by β-arrestin2 in the context of glucocorticoid-induced injury and adrenoceptor modulation in the pancreas is novel.
Pharmacologically, blocking either β or α1-adrenoceptors remarkably mitigated the DXM-induced effects. Notably, β-adrenoceptor blockade largely mediates carvedilol and propranolol effects, and α1-adrenoceptor blockade plays a minor role in mediating carvedilol effects. Combined β- and α1-adrenoceptor blockade produced a superior therapeutic effect over individual receptor targeting, particularly in ameliorating glucose dysregulation, apoptosis, and fibrosis—an approach not previously explored in this context. Interestingly, low-dose phenylephrine yielded modest yet consistent improvement, supporting a context-dependent, potentially protective role of α1-adrenoceptor activation during metabolic stress—a nuance overlooked in prior research.
Furthermore, the shared dependency of carvedilol, phenylephrine, propranolol, and doxazosin on β-arrestin2-linked signaling suggests that this scaffolding protein may be a central regulator of pancreatic remodeling and a promising therapeutic target in IR-associated fibrosis. The study indeed lays a foundation for future translational research in larger, robust studies aimed at addressing pancreatic complications of IR.
Study limitations
This study was exploratory in nature. While it employed a comprehensive pharmacopathological design using multiple adrenergic modulators to investigate the β-arrestin2 crosstalk with cAMP, PKA-ERK1/2-CREB, and PKB-FOXO1 signaling pathways as potential therapeutic targets for DXM-induced pancreatic pathology and provided conceptually novel mechanistic insights, several limitations warrant acknowledgment. First, although robust profiling of apoptotic and fibrotic markers was conducted, direct pathway interrogation using genetic models or pathway-specific inhibitors was beyond the scope of the in vivo design. As such, causality remains to be confirmed in future mechanistic studies. Second, while the Resource Equation Approach suggested a maximum sample size of four rats per group, we used a larger number of animals (n = 9 per group) to account for the anticipated DXM-induced mortality risk. Third, due to resource constraints, sample pooling was used for certain biochemical analyses. This approach, while supported in statistical literature for generating representative group means, precluded assessment of inter-individual variability. However, despite the pooling strategy and small analytical sample sizes, statistical power analysis using G*Power software confirmed that the one-way ANOVA used throughout the study retained strong statistical power (98%–100%). These limitations do not undermine the validity of the reported findings, but rather point to directions for future refinement using molecular interventions and individualized measurements.
Footnotes
Acknowledgements
The authors express their sincere gratitude to King Abdulaziz University in Jeddah, Jeddah University, and all participants who generously agreed to take part in this study.
Author contributions
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Ethical approval and consent to participate
Animal handling was in accordance with the Guide for the Care and Use of Laboratory Animals (8th edition, National Academies Press). The study was approved by the IACUC, which serves as the ethical review board for animal research at Zagazig University (Approval No. ZU-IACUC/3/F/210/2019).
Ethics approval
Ethical approval for this study was obtained from the Institutional Animal Care and Use Committee (IACUC) in Zagazig University (approval No ZU-IACUC/3/F/210/2019).
Animal welfare
The present study followed international, national, and institutional guidelines for humane animal treatment and complied with relevant legislation.
Consent to publish
All authors have read and consented to the publication of this manuscript.
Availability of data and materials
The data that support the findings of this study are available from the corresponding author, Nehal S. Wahba, upon reasonable request.