
Abstract
Immune-checkpoint blockade (ICB) demonstrated inspiring effect and great promise in anti-cancer therapy. However, many obstacles, such as drug resistance and difficulty in patient selection, limited the efficacy of ICB therapy and awaited to be overcome. By timely identification and intervention of the key immune-suppressive promotors in the tumor microenvironment (TME), we may better understand the mechanisms of cancer immune-escape and use novel strategies to enhance the therapeutic effect of ICB. Myeloid-derived suppressor cell (MDSC) is recognized as a major immune suppressor in the TME. In this review, we summarized the roles MDSC played in the cancer context, focusing on its negative biologic functions in ICB therapy, discussed the strategies targeted on MDSC to optimize the diagnosis and therapy process of ICB and improve the efficacy of ICB therapy against malignancies.
Introduction
Immune-checkpoint refers to those inhibitory molecules mainly expressed on immune cells to essentially help prevent immune overreaction. Nevertheless, these pathways may be stolen by tumors to escape the immune surveillance and survive. The best-known co-inhibitory pathways are PD-1/PD-L1, CTLA-4, etc. In recent years, a series of monoclonal antibody drugs (e.g., ipilimumab for CTLA-4; nivolumab and pembrolizumab for PD-1) have been used in clinical practice. These so-called immune-checkpoint blockade (ICB) therapies resulted in surprising outcomes in multiple cancer types.1 -3 However, when clinicians intended to put ICB into more practice, they also encountered numerous difficulties and puzzles. Despite the significant response and survival benefit in a certain group of patients, most patients failed to benefit from ICB therapy. Even those who initially responded well to ICB eventually lost their primary sensitivity to the therapy. In addition to drug resistance, we also lacked definite and concrete criteria to select and determine the appropriate patient subgroup for different ICB therapies, and this depends on the exploration of effective biomarkers to predict ICB efficacy. For example, the factors and molecules, which are able to reflect the TME or the intrinsic immune characteristics of tumor, are the best candidates. 4 Nowadays, PD-L1 expression is the most widely used predictor, which has been studied in multiple clinical trials.5 -7 Generally, high expression of PD-L1 is recognized as a favorable indication for PD-(L)1 blockade. However, the results from different trials are inconsistent, and the cut-off values and scoring systems varied between different trials. 4 Differences in antibody and platform use of immunohistochemistry (IHC), varied reasons for PD-L1 upregulation, and the dynamic changes in the TME may explain the unsatisfied efficacy of PD-L1 expression as a biomarker for ICB efficacy prediction. Moreover, tumor-intrinsic factors, such as tumor mutational burden (TMB), mismatch repair deficiency, microsatellite instability, and oncogenic mutations, have also been investigated for ICB patient selection.8 -10 Although IHC examination of PD-L1 expression,11,12 T-cell infiltration, 13 or genetic testing measuring TMB 14 may indicate valuable reference information for patient selection, the sensitivity, specificity, and accuracy are still far away from expectation. A better understanding of the mechanisms of the emergence of these obstacles and novel strategies to handle these difficulties are necessary to improve the efficacy of ICB and bring better clinical benefits to the people dying of cancer.
Obstacles of immune-checkpoint blockade in anti-cancer therapy
The ICB therapy cuts off the immune-inhibitory pathways and allows T-cell reactivation followed by expansion, recruitment to the TME, antigen presentation, and recognition, which ultimately kills the tumor cells. 15 Besides tumor-killing cytotoxic T cell, effector memory T cell is also indispensable for durable immune protection and disease control.16,17 Generally, an effective anti-tumor response requires sufficient tumor neoantigens, intact neoantigen presentation and recognition, adequate T-cell infiltration and function, and formation of immune memory. 18 However, immune suppressive factors, whether endogenous or exogenous, would impair the steps mentioned above and form immune resistance,16,19 which resulted in ICB therapy failure.
Immune resistance mediated by the TME
The immune resistance derives from dysfunction of T cell, which caused by both tumor-intrinsic and extrinsic factors. With regard to the intrinsic mechanisms of tumor immune evasion, any genetic or epigenetic alterations, which disrupt the proper neoantigen generation, presentation, and key cellular signal pathways, would lead to insufficient T-cell activation and the subsequent immune resistance. On the other hand, the immune cells and stromal cells within the TME also have a tight crosstalk with cancerous cells, which promote the formation of immune resistance and limit the ICB therapy efficacy.
Diversified immune cells and inflammatory mediators constitute the TME. The best-known participators include tumor-infiltrating lymphocytes (TILs), cancer-associated fibroblasts (CAFs), tumor-associated macrophages (TAMs), tumor-associated neutrophils (TANs), dendritic cells (DCs), MDSCs, etc.20,21 Various functions exerted by these cells come to the same result: immune suppression. Here, we focus on MDSC, a potent population of immune suppressor cells, in the TME, discuss its immune suppressive mechanisms, and address the possibilities of utilizing novel therapeutic strategies targeting on MDSC to enhance ICB therapy efficacy.
MDSC: Major regulators of tumor immune suppression
Myeloid-derived suppressor cells (MDSCs) in short represent a heterogeneous population of immune suppressive myeloid cells, which are abnormally expressed in multiple disease settings, such as autoimmune diseases and cancer. 22 The primary identification of MDSC dated back to the 1980s, when researchers found there existed a group of suppressor cells, which inhibit the T-cell proliferation and function in mice. 22 In the following years, several studies confirmed the existence of this cell population and demonstrated their immune inhibition function in different types of cancer context.23,24 With numerous investigations and unremitting efforts by the scientists, this intriguing cell population was finally termed as “myeloid-derived suppressor cell” in 2007, 25 which opened up a new world for immunology research field.
MDSC, primarily defined as CD11b+ Gr-1+ cell, is heterogeneous and can be further divided into two subgroups: polymorphonuclear MDSC (PMN-MDSC) and monocytic (M-MDSC). 26 In humans, PMN-MDSC is defined by the expression of markers of CD11b+CD14−CD15+ or CD11b+CD14−CD66b+ and M-MDSC as CD11b+CD14+HLA-DR−/loCD15−.27,28 Similar to human MDSC, murine MDSC definition also depends on co-expression signature of a series of biomarkers. Murine M-MDSC is defined as CD11b+Ly6C+Ly6Glow/-, whereas PMN-MDSC is classified as CD11b+Ly6C−Ly6G+. 26 Nevertheless, the heterogeneity of MDSC is not only limited to the differentiation of its marker expression; different tumor type, burden, anatomical position along with the distinct soluble factors, and cellular communication made the identification of MDSC subsets even more complicated.
Characterized by its immune suppression feature, MDSC remains a major inhibitor of T lymphocyte activation (as shown in Figure 1). Multiple effector molecules and signaling pathways are used by MDSC to regulate immune suppression. The main mechanisms involve depletion of necessary amino acids, production of nitric oxide (NO), reactive oxygen species (ROS), transforming growth factor-β (TGF-β), and other cytokines, PD-L1 expression, downregulation of L- and E-selectins and so on. 22 In addition, MDSC can also impair T-cell function indirectly by induction of other immune-inhibitory cells, such as regulatory T cells (Tregs)29,30 and Th17 cells.31,32 The exact immune-suppressive mechanism used by MDSC varies according to many factors, such as MDSC phenotype, cancer type and stage, and the disease progression.33,34

MDSC-regulated immunosuppression in cancer following ICB treatment. ICB treatment inhibited the pathways used by tumor to escape the immune attack by T cell and thus reinvigorated the tumor-killing capabilities. However, MDSC, which differentiated from hematopoietic stem cells (HSCs) in the bone marrow, would expand and be recruited to the tumor site. MDSC suppressed the immune responses through multiple mechanisms, which subsequently impaired the efficacy of ICB treatment and formed immune resistance. ICB: immune-checkpoint blockade; MDSC: myeloid-derived suppressor cell.
MDSC accumulation and recruitment in ICB therapy
Upregulation of MDSC expression in peripheral blood, lymphoid tissue, and tumor site has been found to be significantly correlated with unfavorable outcomes and short survival in patients with multiple cancer types.35 -37 The increasing numbers of MDSC subsets mainly derive from two distinct routes: accumulation in quantities and spatial migration to the TME.
Accumulation of MDSC depends on complex mechanisms. Condamine et al 38 addressed this phenomenon by using a model to divide the accumulation process into two different but partially overlapping signaling pathways: one is by inhibiting the immature myeloid cells normal differentiation, whereas the other entails the conversion from immature myeloid cells into MDSC on detrimental abnormal activation. The network of transcriptional factors regulating MDSC accumulation has been depicted in detail by a series of studies. On activation by upstream factors, STAT3, IRF8, and other transcriptional regulators could positively or negatively mediate the expansion of MDSC. △NP63, an isoform of transcription factor p63, has recently been found to induce MDSC recruitment through activation of CXCL2 and CCL22 signaling pathways in triple-negative breast cancer, 39 which subsequently promoted tumor progression. In addition, Liang et al 40 demonstrated that stimulator of interferon genes (STING) activation resulted in MDSC infiltration in the TME in resistance to radiation therapy. However, whether STING mediate immunosuppression during ICB therapy is still unknown to us. Recently, c-Rel, a member of NF-κB family, is found to be involved in the regulation of MDSC differentiation by directly promoting Cebpb expression. Furthermore, a combination of c-Rel inhibitor and PD-1 blockade significantly boosts anti-cancer response by CD8+ T cells. 41
In addition to the accumulation of MDSC regulated by transcriptional pathways, MDSC can also be recruited to the tumor tissue by the interaction between chemokines, cytokines, and their receptors, 42 which help to constitute the TME and promote tumor progression. These chemokines include CXCL, CCL, and CX3CL, CSF families and are mostly produced by tumor cells, tumor-associated immune cells and stromal cells in the TME. MDSC is also found to be a chemokine source in the TME. Holmgaard et al 43 demonstrated that MDSC showed an increased expression following CTLA-4 blockade in tumor-bearing mice, and a timely inhibition of CSF-1/CSF-1R pathway significantly improved anti-CTLA-4 ICB treatment by depleting tumor-infiltrating MDSC. This study indicated that targeting CSF-1/CSF-1R could be selected as a valuable therapeutic strategy with multiple potential benefits for reversing ICB resistance and optimizing the efficacy of immunotherapy. First, blockade of CSF-1/CSF-1R possessed great specificity. Previous studies found that CSF-1R was mainly sourced from myeloid cells in the TME, including MDSC43,44 and TAM, 45 depending on the distinct tumor type. It also reminded us that the dominant tumor-infiltrated myeloid cell subtype in the specific tumor type need to be figured out before choosing CSF-1/CSF-1R blockade. Second, the regulation of MDSC via CSF-1/CSF-1R blockade is modulated by multiple pathways, which endows CSF-1/CSF-1R blockade with greater efficacy. Some studies proved that CSF-1R inhibition could decrease the numbers of MDSC,45,46 whereas others indicated that CSF-1R blockade could favorably reprogram MDSC and induce an alteration of MDSC toward an antitumor phenotype. 47 Of note, CSF-1R blockade could regulate both the number and function of MDSC in breast cancer. 43 Third, upregulation of immunosuppressive cells and molecules is a common immune-evasive mechanism during ICB treatment, which forms a positive feedback loop to impair immune responses. 48 CSF-1R expression on MDSC was found to be enhanced during CTLA-4 treatment, which resulted in acquired immune resistance against ICB treatment. 43 Therefore, the CSF-1/CSF-1R blockade in combinative use with ICB treatment could exert synergic effect and improve antitumor responses. Similarly, IL-10 is produced by tumor-infiltrating myeloid DCs to promote MDSC accumulation in the TME as an adaptive immune resistance against PD-1 blockade. 49 Based on different mechanisms that MDSC use to accumulate in cancers, researchers have developed various therapies and drugs targeted on certain upstream factors and receptors to reduce MDSC and potentiate ICB efficacy. Although multiple chemokine-targeted therapies against MDSC have been proved to harness greater response in combination with ICB, one critical issue that cannot be neglected is that most of these MDSC-recruiting chemokines also target other effector immune cells, including T cell,50 -52 NK cell, 53 and other tumor-killing immune cells. Therefore, both positive and negative sides of chemokine blockade require careful consideration before use and more specific and effective targets are urgently needed. Semaphorin 4D (Sema4D; CD100), traditionally recognized as a tumor-promoter gene, has recently attracted wide attention for its immune-regulatory roles, especially about MDSC. In human head and neck squamous cell carcinoma, sema4D has a similar function to GM-CSF in inducing MDSC expansion. 54 What’s intriguing is that sema4D also mediates MAPK-dependent chemokine release from tumor cells, which promote MDSC recruitment and activation. 55 However, the exact mechanism of sema4D-MDSC interaction is still elusive. Further studies are needed to determine the receptor which sema4D binds with in the TME and the downstream signaling pathways. Furthermore, another regulator of MDSC called CD200 is also found to mediate immunosuppression in pancreatic ductal adenocarcinoma. 56 Although single-cell RNA sequence uncovered several suspicious genes involved in CD200/CD200R axis, the definite pathway remains a riddle.
The microRNA (miRNA), a member of non-coding RNA family, represents a small RNA molecule containing approximately 22 nucleotides. By regulating post-transcription of gene expression, miRNA mediates gene silencing and involves in multiple biological processes, including cancer development. 57 Finally, miRNA has also been identified to participate in MDSC differentiation. For instance, tumor-derived miR-30a directly inhibits suppressor of cytokine signaling 3 (SOCS3) and induces MDSC expansion and activation through JAK/STAT3 pathway in B-cell lymphoma. 58 Moreover, in gastric cancer, researchers found miR-107 could be capsuled in exosomes delivered by tumor cells that promote MDSC accumulation and function by targeting PI3K pathway. 59 Due to the wide and powerful regulatory roles played by miRNAs on MDSC, targeting MDSC-related miRNAs has been studied as a potential therapeutic for cancer immunotherapy. For example, Wang et al 60 proposed that miR-155 served as an important tumor suppressor in melanoma and lung cancer by inhibiting MDSC recruitment through hypoxia inducible factor (HIF)-1α blockade, which decreased the secretion of MDSC-recruiting factors. More importantly, miR-155 deficiency could result in a more immunosuppressive phenotype of MDSC with more iNOS and Arginase 1 (ARG-1) production. This study showed that miRNAs could not only target on the upstream factors of MDSC expansion and recruitment but also directly regulate the immune functions of MDSC. However, miR-155 was oppositely found to be able to synergize with miR-21 to promote MDSC expansion through SHIP-1/STAT3 pathway. 61 In line with this study, blockade of miR-155 could improve the killing capacities of T cells via inhibition of MDSC function. 62 Furthermore, a range of miRNAs have been discovered to participate in the conversion and infiltration of MDSC and strongly associated with the immune resistance against ICB treatment. 63 Despite large amounts of studies have been done to elucidate to role of miRNAs in the process of MDSC regulation and ICB treatment, the exact mechanism remains elusive mostly due to the heterogeneity of cancer and the factors influencing the imbalance of MDSC-related miRNAs. In a word, MDSC-related miRNA has demonstrated promising potential in predicting immunotherapy outcome and harnessing ICB efficacy, but still more studies are required for future clinical practice.
MDSC-Mediated Immune Resistance in ICB Therapy
As mentioned above, MDSC exert immunosuppressive functions, which could be hijacked by tumor to mediate immune resistance against immunotherapy. Understanding the mechanisms used by MDSC for immunosuppression may help us develop more strategies to reactivate anti-cancer immune responses and improve ICB effect.
The TME-derived chronic inflammatory mediators stimulate immature myeloid cells and promote them to differentiate into MDSC circulated in peripheral blood and finally infiltrate the tumor. A common well-known function shared by both subsets of MDSC depends on arginase production, which deprives T cell of amino acid arginine and ultimately inhibits T-cell proliferation. 64 ARG-1 is released in the tumor milieu by MDSC to create an immunosuppressive TME by depletion of normal TCR function.65,66 Targeting on the major immune inhibitory role played by ARG-1, multiple studies have been made to explore the efficacy of ARG-1 blockade in cancer therapy. Romano et al 67 found that in multiple myeloma, increased expression PMN-MDSCs exert crucial immunosuppressive roles through ARG-1 production, and the administration of ARG-1 inhibitor nor NOHA could increase the effects of lenalidomide or bortezomib treatment. In addition, another study found the administration of Brentuximab Vedotin as single agent in Hodgkin lymphoma could reshape the immune microenvironment by reducing the amounts of MDSC and ARG-1 production, which finally improved the survival of Hodgkin lymphoma patients. 68 In all, ARG-1, an enzyme produced by MDSC in the TME, depleted the arginine and thus inhibited the T-cell killing function and caused immune escape. Targeting on ARG-1 has been proposed as a potential effective method to trigger immune responses and helped reverse immune resistance. However, relative clinical trials of monotherapy or combinative therapy with ICB are still elusive. Studies also indicate cysteine, which is necessary for T-cell activation,69,70 could be sequestered by MDSC from the TME, further weakening anti-cancer immunity. 71 Notably, richness in cysteine could be used as a biomarker in combination with protein acidic secretion to evaluate MDSC suppressive degree. 72 In addition, tryptophan level could also be reduced by MDSC through indole amine 2,3 dioxygenase (IDO) production.73,74 In addition, IDO could mediate immunosuppression by production of cytotoxic substances to repress Ag-specific immune responses and induce Treg production.75 -77 An IDO inhibitor 1-methyl-L-tryptophan or STAT3 antagonist JSI-124 could thus block the immune suppressive activity of MDSC and serve as an efficient approach to optimizing ICB efficacy. 78 In addition, Qu et al 79 proposed that blockade of CARD9-NF-κB-IDO pathway helped inhibited the suppressive function of MDSC to inhibit the progression of colon cancer. Of note, IDO pathway is not only an immunosuppressive mechanism used by MDSC, but also an upstream factor to recruit and activate MDSC. Holmgaard et al 80 found that tumor could express IDO to activate Tregs, which subsequently promoted MDSC migration to the tumor site to enhance tumor progression as well as induce immune resistance to ICB. These findings suggested a major linking network between MDSC and tumor mediated by IDO, and therapeutic targeting IDO could develop great potential. Nevertheless, Feng et al 81 demonstrated a novel mechanism used by MDSC to influence T-cell function; a crucial component protein of T-cell receptor signaling pathway named lymphocyte-specific protein tyrosine kinase (LCK) could be nitrated by MDSC, which impaired T-cell function and limited ICB efficacy in prostate cancer. Moreover, MDSC is a powerful source of anti-inflammatory mediators. Researchers identified that tumor-infiltrated MDSCs produce TGF-β in an autocrine feedback loop, which inhibits cytotoxic T-cell proliferation and induces Treg accumulation. 82 IL-6, a pivotal immune suppressor in cancer, has also been identified as a functional mediator released by MDSC to promote tumor aggressiveness and stemness. 83
We discussed above that MDSC differentiation and activation could be mediated by cell-to-cell communication via exosome transport of biological small molecules. In turn, MDSC may also exploit this mechanism to exert immune suppressive functions. Geis-Asteggiante et al 84 established a 4T1 tumor system model in mice to obtain MDSC under “conventional” and “inflammatory” conditions. Afterwards, the exosomes derived from MDSC and the cargoes of mRNAs, miRNAs or proteins were intensively studied. Evidence indicate MDSC-derived small RNA molecules may involve in the immune suppressive activity. MDSC is found to secret miR-126-contained exosomes under doxorubicin chemotherapy to form acquired chemo-resistance. 85 In addition to cancer, MDSC-derived non-coding RNAs also participate in the immune regulation in other disease settings like arthritis 86 and lupus. 87 However, whether and how MDSC-derived exosomes mediate immune resistance in ICB therapy remain to be elucidated.
Resistance to ICB treatment could be categorized into two main types: primary resistance and acquired resistance. Depending on their powerful immunosuppressive capacities, MDSC played a major role in the regulation of both primary and acquired resistance in different cancer types. Primary resistance referred to the low or non-response to ICB therapy. Many factors, such as low PD-L1 expression88,89 and insufficient tumor mutational burdens and neoantigen presentation90,91 and even microbiome92,93 were found to impact the ICB efficacy and form the primary resistance. Emerging evidence indicated that MDSC is also an important participator in the formation of primary resistance. Meyer et al 94 proposed that low frequencies of circulating MDSC in the anti-CTLA-4 treatment represents a low resistance to ICB and predicts a better outcome in the metastatic melanoma patients. In line with the results of this study, another study confirmed that in advanced melanoma, an upregulated level of circulating MDSC is strongly associated with the absence of melanoma antigen-specific T cells and a poor survival of melanoma patients. 95 These studies indicated that a primary high expression of MDSC in the TME may helped promote the primary resistance via the formation of an immunosuppressive milieu, However, the primary resistance varied significantly between cancer types and individuals, 96 and the mechanisms used by MDSC to mediate primary are still unclear. Therefore, further investigations into this issue are required. Unlike primary resistance, acquired resistance represents a selection or evolution of tumor cells, which develop mutations in critical pathways in ICB. Alterations of MDSC accumulation and function could join this process of modulation and enhance the acquired immune resistance. Limagne et al 97 found that MDSC impaired the cytotoxic functions of CD8+ T cells by expressing galectin-9 to interact with TIM-3 on T cells, which mediated both the primary and acquired immune resistance to anti-PD-1 therapy. In addition to the MDSC function, another study suggested that in response to PD-1 blockade, a PD-L1/NLRP3 inflammasome signaling cascade pathway was activated, which subsequently recruited large amounts of PMN-MDSC to the tumor site and dampened the antitumor immune responses. 98 In agreement with this study, multiple studies also indicated that accumulation of MDSC in the TME influenced the efficacy of immunotherapy agents.99 -101 To better understand the role played by MDSC in mediating acquired immune resistance, we need to perform more investigations on the dynamic changes of the status of MDSC during the process of ICB treatment. The mechanisms used by MDSC to influence ICB efficacy are summarized in the Table 1.
Mechanisms used by MDSC to regulate immune resistance to ICB.
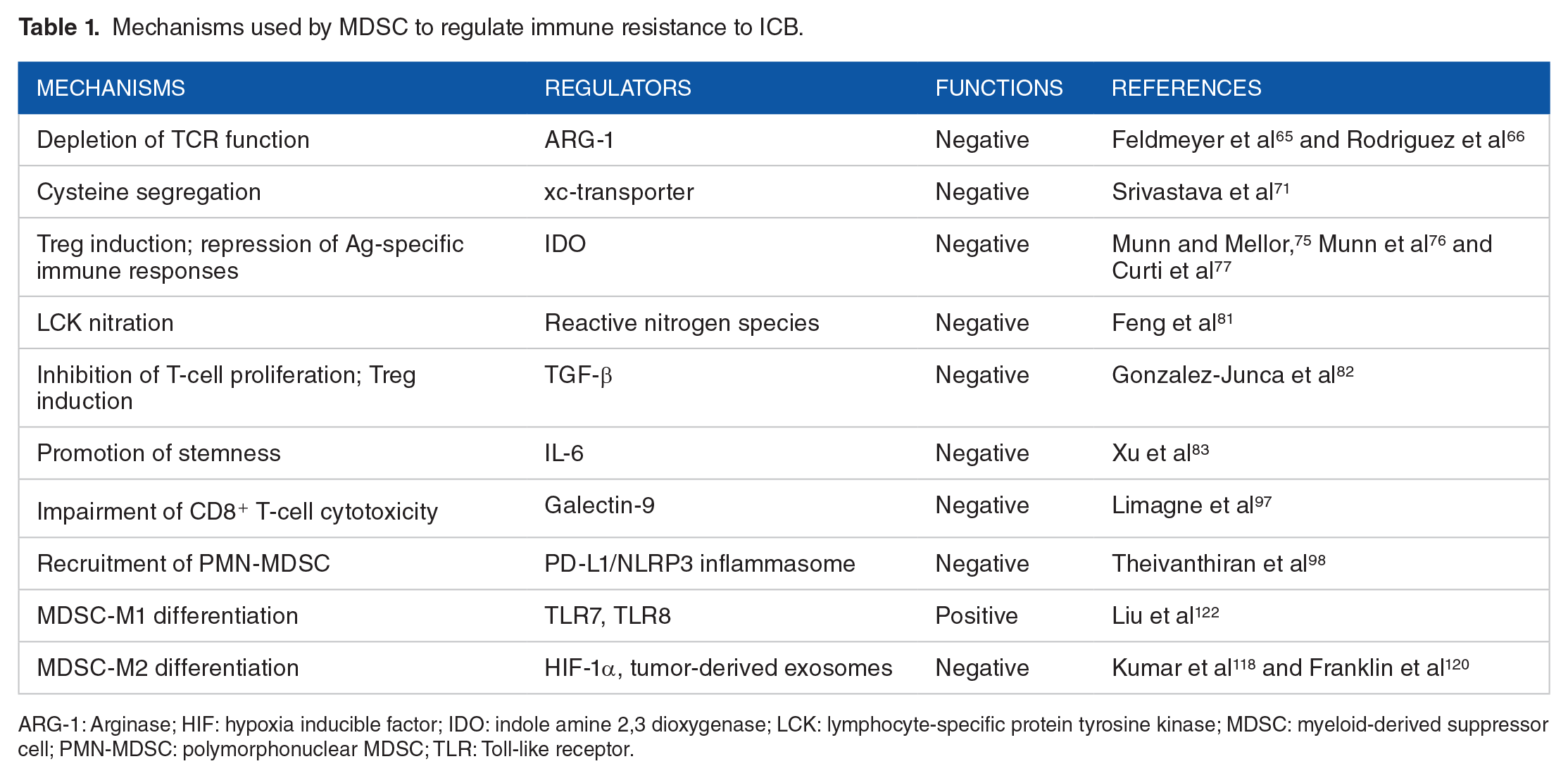
ARG-1: Arginase; HIF: hypoxia inducible factor; IDO: indole amine 2,3 dioxygenase; LCK: lymphocyte-specific protein tyrosine kinase; MDSC: myeloid-derived suppressor cell; PMN-MDSC: polymorphonuclear MDSC; TLR: Toll-like receptor.
The Interactions Between MDSC and Other Components in the TME
The TME constitutes a great shelter for tumor cells and other components to either survive or interact with each other constantly, which favors tumor progression. Recently, growing evidence indicates that the external signaling network scattered between various immune cells and cancer cells foster tumor immune resistance and relapse besides cancer cells themselves. MDSC, the major immune suppressor in cancer, is no exception and has a tight and complex crosstalk with other components in the TME.
CAFs are identified as a dominant stromal component in the TME, and they modulate tumor progression through numerous mechanisms, such as micro-environment remodeling, angiogenesis induction, and immunosuppressive functions.102 -104 Recently, growing evidence suggest that CAFs also interact with MDSC intimately to regulate immune responses indirectly in the TME. CXCL family, as discussed above in the second section, represents a major chemokine for MDSC recruitment to the tumor site. Kumar et al 105 found that CAF could produce CXCL2 to promote PMN-MDSC migration to the TME, which forms a resistance against anti-CSF1 mono-antibody therapy. Likewise, Sano et al 106 demonstrated that CXCLs-CXCR2 signaling pathway could be used by CAFs to maintain the chronic inflammatory micro-environment, including MDSC infiltration. Furthermore, CCL2-CXCR2 axis also mediate MDSC recruitment in the TME of lung squamous cell cancer and induce immunosuppression via ROS production. 107 These studies remind us with the importance of identifying upstream regulatory factors of MDSC migration in the TME from distinct cells and cancer types, and novel therapeutic strategies against MDSC in ICB could target on CAF or other accessories in the TME. For example, the efficacy of an FAPα/surviving-targeted DNA vaccine, which depleted CAF in the TME, was found to be enhanced in combined use with MDSC elimination. 108
Owing to relatively low expression and short lifespan, PMN-MDSC is not so well-studied in the TME, and M-MDSCs have been viewed to represent the major immunosuppressive myeloid cells in the TME.34,109 Notably, M-MDSCs have high plasticity and is able to differentiate into multiple types of other myeloid cells in the TME. 34 TAMs refer to the macrophages infiltrated within the TME and are usually tamed to acquire pro-tumoral properties.110 -112 Based on the activation state and functional characteristics, TAMs can be divided into two subgroups: tumor-killing M1 phenotype and tumor-promoting M2 phenotype. 113 Accordingly, the M1/M2 ratio can serve as an effective biomarker to predict tumor progression114,115 and resistance to immunotherapy.116,117 Numerous studies have described that MDSCs could differentiate into TAMs in the TME.118 -120 Therefore, the dual-direction conversion from MDSC to macrophage in the TME may also mediate immune responses not to be ignored. Biswas et al 121 described that exosomes released by mesenchymal stem cells (MSCs) promote MDSC differentiation into M2-like TAMs in the breast cancer, which significantly impaired anti-cancer immunity. Recently, Toll-like receptors (TLRs) are found to regulate MDSC to differentiate into M1 macrophages, 122 which indicates a novel pathway to block the immune suppressive functions of MDSC and harness the efficacy of ICB therapy. Factors controlling the transformation from MDSC could be divided into two aspects. For one thing, the distinct hallmarks of TME are major driving forces for MDSC differentiation into pro-tumor M2 macrophages, such as hypoxia 118 and intercellular interactions via extracellular vesicles, 120 which reminded us that we need to figure out the exact mechanisms and pathways used by tumor to divert MDSC into more immunosuppressive macrophages. For another thing, there are also potential factors that are able to convert MDSC into anti-cancer M1 type macrophages. For example, TLR7/8 activation 122 or phosphatidylserine blockade 123 successfully polarized MDSC into M1 macrophage, which reactivated antitumor immunity. Whether these factors participated in the formation of immune resistance to ICB is still elusive. Therefore, further investigations into this issue are required. However, in addition to the transformation relationship between MDSC and TAM, their direct mutual interaction within the TME has not been well explained. Besides macrophage, other mature immune cells may also exert crucial immune functions in contact with MDSC. Neutrophils are traditionally identified as the basic defense line of immune system against infection. Nevertheless, on certain stimuli, such us ROS or granule-derived myeloperoxidase (MPO) production, mature neutrophils could form MDSC-like properties and inhibit T-cell function in the cancer context. 124 In addition, MDSC is found to be able to differentiate into DCs and even fibrocytes. 125 Up until now, how the interactions between MDSC and other components in the TME participate in the formation of immune resistance to ICB is still unclear to us. Therefore, more studies are needed to expand this domain for ICB synergism.
Gut microbiota refers to trillions of microbes residing in the mammalian gastrointestinal tract and co-evolving with the host. This symbiotic relationship helps maintain the systemic homeostasis. Recently, growing evidence suggests that gut microbiota exert key roles in the formation of innate and adaptive immunity and participate in the process of carcinogenesis. 126 In addition, alterations in gut microbiota have significant impacts on the efficacy of cancer immunotherapy.127,128 Remarkably, MDSC also have intimate interactions with gut microbiota. Dong et al 129 demonstrated that a gut symbiotic bacteria named Fusobacterium nucleatum (Fn), specifically promotes MDSC accumulation in the TME and subsequently inhibits the anti-cancer immune responses, and this negative effect could be reversed by total elimination of Fn via treatment of a nanoparticle-assembled phage. Surprisingly, gut microbiota is found not to only settle in gastrointestinal tract but also may migrate to other anatomical sites to modulate immune functions. For example, a distinct group of microbes are found to transmigrate from gut to pancreas to entail immunosuppression by TME reprogramming, which assists pancreatic tumor progression. Correspondingly, the depletion of bacteria synergizes with anti-PD-1 ICB treatment via inhibition of MDSC expansion and reactivation of T-cell function. 130 These studies revealed the potential therapeutic value of microbiome in ICB treatment, but the molecular mechanisms still require further clarification. Although quite a few commensal bacteria have been identified to promote MDSC accumulation during tumor progression, another study indicates that the early-life exposure to flora from external environment is indispensable for the formation of an intact immune system by PMN-MDSC restriction and inhibition of CXCL expression. 131 We can see from it that gut microbiota may serve as a double-edged sword in the regulation of MDSC function. On the other hand, fungi, which accounts for less than bacteria in the gut microbiota and are often neglected by us, may also represent a pivotal subgroup of host microbiome to mediate anti-cancer immunity. Wang et al 132 described abnormal overexpression of fungi in the gut, especially Candida Tropicalis, would promote progression of colitis-associated colon cancer by induction of MDSC accumulation. Although the exact mechanisms remain unclear, several studies could serve as valuable references for us to speculate how gut microbiome promoted the accumulation of MDSC. Recently, Zhang et al 133 demonstrated that in the mice models of Cholangiocarcinoma, an impairment of gut barrier could result in the migration of gut-derived bacteria to the liver, which induced CXCL1 secretion from hepatocytes through TLR4 pathway and subsequently caused PMN-MDSC accumulation. This means that the gut microbiota could serve as pathogen-associated molecular patterns to interact with the pattern-related receptors and create an immunosuppressive environment in the target organ through induction of MDSC-recruiting cytokines and chemokines. However, this process required the incompleteness of gut barrier and the escape and migration of gut microbiota. In addition, a novel study pointed out that intestinal DCs could serve as vehicles to transport the gut microbiota-derived antigen into the thymus to educate the thymic T cells and form and strengthen the anti-infective capacities of the host. 134 Based on this brilliant study, we may naturally make a hypothesis that under cancer conditions, the altered gut microbiota-related antigen may also arrive at important immune organs, such as tumor-draining lymph nodes, bone marrow, and liver in this way and regulate the recruitment, accumulation, and functions of various immune cells, including MDSC. However, these hypotheses required future experiments and clinical trials to confirm. Strictly speaking, the host microbiome may not belong to TME components. However, accumulating evidence indicates that gut microbiota is a major participant in the innate and adaptive immune system and influence cancer development in a both direct and remote modulatory manner. Therefore, the interactions between MDSC and gut microbiota need to be elucidated more in detail. A breif overview of pathways of MDSC accumulation, migration and functions in the TME is shown in Figure 2.

An overview of pathways of myeloid-derived suppressor cell (MDSC) accumulation, migration, and functions in the tumor microenvironment (TME). Various cytokines and chemokines derived from the TME induce the expansion and accumulation of MDSC and facilitate its migration into tumor site, which led to inhibition of tumor-killing T cells and NK cells with various suppressive factors. Meanwhile, intimate interactions existed between MDSCs and other components in the TME.
Targeting MDSC: New Therapeutic Strategies and Insights for ICB Anti-cancer Therapy
Combination of systemic chemotherapy with ICB treatment
Despite the constant breakthroughs of ICB practice in cancer treatment, traditional systemic chemotherapy remains the first-line standard treatment for most malignancies. With the development of ICB agent application, clinicians and researchers have long been questioning whether combined use of ICB and chemotherapy could bring the patients greater survival benefits. Therefore, numerous clinical trials have been conducted to verify this hypothesis.135 -137 Surprisingly, increased efficacy and good security were obtained by the combination strategy. However, the mechanism for the synergism remains unsolved. Some studies suggest that chemotherapy could induce immunogenic cell death (ICD), which causes exposure of tumor antigen and activates cytotoxic T cells, DCs, and macrophages.138,139 Moreover, chemotherapy is also found to be able to regulate TME and act on multiple immune inhibitory cells, including MDSC.
Cisplatin is one of the most common cancer chemotherapeutic agents for a wide range of cancers, including lung cancer, bladder cancer, breast cancer, etc., depending on its ability to induce cell death by inhibiting DNA replication and transcription.140,141 With the development of ICB therapy, the immune-regulatory roles of cisplatin attracted much attention. Wu et al 142 investigated the immune effects of low-dose cisplatin treatment on bladder cancer and found that cisplatin could specifically eliminates periphery PMN-MDSC and thus enhances CD8+ T-cell function. Similar effects have also been detected in melanoma 143 and ovarian cancer. 144 Interestingly, the inhibitory impact on MDSC from cisplatin seems not only limited on periphery blood but also on other immune organs. For example, cisplatin is found to significantly reduce splenic MDSC and promote IFN-γ-produced myeloid cells expansion in metastatic breast cancer model. 145 Conversely, MDSC could also participate in the formation of cisplatin-resistance of cancer, 146 which suggests the necessity of combination use of ICB and MDSC-targeted drugs in face of cisplatin-resistant cancer patients. Oxaliplatin is one of the third-generation platinum-based chemotherapeutic drugs, which owns lower adverse toxicity and shows similar performances in several cancers compared with cisplatin.147 -150 Kim et al 151 demonstrated that oxaliplatin selectively depletes M-MDSCs and promotes MDSC maturation in colorectal cancer models by modulating NF-κB signaling pathway. Hence, new strategies of combination of oxaliplatin and ICB may obtain great responses in cancer treatment after more clinical trial verification. Nevertheless, under certain circumstances, chemotherapy may also exert undesired immune-regulatory functions. Gemcitabine, which was identified to deplete MDSC in vivo, 152 has recently been found to promote M-MDSC migration into tumor site through upexpression of GM-CSF in breast cancer after repeated utilization. 153 This phenomenon highlights the importance of quantity determination when we formulate chemotherapy–ICB combination strategy. Although a range of studies have demonstrated the immune-regulatory function of MDSC depletion by chemotherapy, we still wonder whether this function could be used to obtain an optimized efficacy of chemoimmunotherapy through MDSC depletion. Combinative use of cisplatin, anti-PD-1 antibody, and pemetrexed was found to enhance the antitumor immune responses in mesothelioma mice at least in part due to the decrease of MDSC infiltration. 154 Another study indicated that in HPV-related cancers, cisplatin was able to sustain an induction of tumor-specific CD8+ T cells along with a decrease of CD11b+GR-1int myeloid cells, and a combination with anti-CTLA4 helped improve the efficacy. 155 As discussed above, chemotherapy could induce ICD, which partly explained the enhanced efficacy of combinative administration of chemotherapy and immunotherapy. However, ICD is relative difficult to quantify in vivo. Thus, immunosuppressive cells received a substantial focus. From previous studies, we could conclude that the immunopotentiation function mediated by chemotherapy is in versatile targets, and MDSC depletion served as an important mechanism to participate in this synergistic effect.
Molecule-targeted agents for MDSC inhibition
According to the molecular basis of MDSC recruitment and expansion, many involved signaling pathways and key regulatory mediators could be targeted to inhibit MDSC accumulation and harness ICB efficacy. As has been discussed above, a lot of chemokines interact with corresponding receptors expressed on MDSCs and promote their tumor trafficking. By using CXCR2 antibody, researchers successfully disrupted MDSC tumor infiltration and significantly increased anti-PD-1 immunotherapy efficacy.156,157 As expected, targeted blockade of CXCL12/CXCR4 obtained similar survival benefits in combination with PD-1/PD-L1 axis blockade in ovarian cancer. 158 However, the specificity of chemokine blockade is still far from satisfying owing to its potential negative effects on other immune cells along with unpredictable side effects.
Due to the irreparable defects of chemokine blockade, more specific targets are urgently desired by clinicians to handle the immune escape mediated by MDSC in ICB treatment practice. Sema4D has been proved to be able to recruit MDSC to tumor site and disrupt T-cell function. 54 And targeted inhibition of sema4D resulted in enhanced efficacy of ICB treatment. 55 Nowadays, the clinical trial of sema4D-based molecule targeted therapy is underway, and we all look forward to its results. Furthermore, CD200 blockade also improves PD-1 antibody therapy, which indicates a novel target for MDSC inhibition. 56 Cell cycle-related kinase (CCRK) belongs to cyclin-dependent kinases (CDK) family, which exerts crucial functions in regulation of cell cycle and transcription. 159 Zhou et al 160 addressed the relationship between MDSC and CCRK in hepatocellular carcinoma and found that inhibition of CCRK successfully depleted MDSC and enhanced ICB efficacy.
A crucial issue that we cannot neglect is the prioritization strategies for us to take when we face so many targets and agents for combinative use. Firstly, we should take the tumor heterogeneity into consideration. A simple but valuable classification framework of TME stratification was set to help us select ICB treatment strategies. Based on the presence or absence of TILs and PD-L1 expression, the TME was classified into four subtypes: Type I: PD-L1 positive with TILs, indicating a formation of acquired immune resistance; Type II: PD-L1 negative with no TILs infiltration, indicating the immune ignorance; Type III: PD-L1 positive with no TIL, indicating an intrinsic formation of immunosuppression; and Type IV: PD-L1 negative with TILs infiltration, indicating an immune escape mediated by other pathways. 161 Different tumor types correspond to these TME types to guide ICB administration. 162 Four aspects would be covered when we perform the combinative therapeutic strategy with ICB, including reversing tumor immunosuppression, induction of immunogenic cell death, enhancement of antigen presentation, and sustainment and activation of cytotoxic T cells. Therefore, the prioritization strategy should be based on the tumor immune status during ICB treatment. We have discussed above the various mechanisms taken by MDSC to mediate the suppression of antitumor immunity. Thus, in the TME of types II and IV, targeting against the MDSC-related cytokines and chemokines may be the first choice to synergize with ICB treatment. Multiple preclinical and clinical trials found that blockade of CSF-1R,163,164 CCL2,165,166 and other factors 167 could enhance the efficacy of ICB. As with the type I TME, which has formed acquired immune resistance to ICB, agents targeting MDSC-secreted mediators may be more effective, such as ARG-1, iNOS, and IDO, which could reverse the immunosuppressive TME and reactivate the previously restricted T cells. However, up until now, there still lakes a consensus of the polarization strategy for the combinative therapy with ICB. Ongoing preclinical and clinical trials will gradually solve the mystery.
Epigenetic drugs for MDSC regulation and ICB efficacy enhancement
Recently, epigenetic dysregulation in the tumor biology has attracted increasing attraction for its broad participation in almost all hallmarks of cancer.168,169 Based on this, many novel epigenetic drugs have been investigated to reverse immune resistance and synergize with ICB treatment. And MDSC is also an important target for epigenetic modulatory drugs.
Histone acetylation helps modify chromatin organization and regulate gene expression. 170 Targeting on this crucial epigenetic regulatory mechanism, histone deacetylase inhibitors (HDACs) has been recognized as a potential candidate agent for MDSC modulation. Valproic acid (VPA), an HDACi-targeting HDAC1, 2, and 3, was found to be able to impair the immunosuppressive functions of MDSC. 171 Furthermore, Youn et al 172 found a critical role of HDAC-2 to hinder monocyte from differentiating into DCs and macrophages but PMN-MDSC, which could be reversed by VPA treatment. Based on the previous findings, Adeshakin et al 173 combined PD-L1 blockade and VPA to evaluate whether immune regulation of MDSC by VPA could synergize with ICB in tumor-bearing mice and found that the immunosuppressive capacities of MDSC were significantly attenuated along with a stronger antitumor immune response and slower tumor progression. Similar results have been found in other HDACi drugs, such as CG-745, 174 ACY-241, 175 and MS-275. 176 Nevertheless, most of these studies are preclinical trials, and we still need more clinical trials in the future to evaluate the value of HDACi drugs in ICB treatment.
In addition to histone acetylation, DNA methylation is another important epigenetic modulatory way in cancer. Recently, Saleh et al 177 found that the DNA methylation is one of the key epigenetic participators in the modulation of multiple inhibitory and immunosuppressive genes of MDSCs, which suggest that the DNA methylation may also be a potential epigenetic target to regulate MDSC functions. Sido et al 178 proposed that an exogenous cannabinoid derived from the Cannabis sativa plant could modulate the DNA methylation of various genes, which were involved in the functions of MDSC including ARG-1 and STAT3. Recently, an expressional profile analysis of m6A RNA methylation regulators in renal cancer suggested that a risk-scoring system based on the levels of m6A RNA methylation could predict the status of immune evasion and serve as an independent prognostic marker. 179 Hopefully, more effective epigenetic drugs will enter clinical trials and help improve the efficacy of ICB treatment.
Designing and synthesis of novel drugs
Nanoparticles have achieved great advances over the last decade as a novel drug delivery system, which helps increase drug biological activity, lower drug toxicity, and promote the drug accumulation in the desired targeted site. Under the circumstances of immune resistance against ICB, nanoparticle have potentials in remodeling TME and boost immunotherapy. Zhang et al 180 enclosed the PARP inhibitor talazoparib in a nano-liposome and evaluated its efficacy on BRCA-deficient breast cancer. Results showed that besides a significant decrease in side effect, the nano-talazoparib treatment dramatically deplete the MDSC, both in tumors and spleens. Furthermore, the formulation of a Gemcitabine nanoparticle also successfully reduced immunosuppression by eliminating MDSC and Tregs in melanoma, which ultimately reactivate T-cell immune responses. 181 In addition to delivery of chemotherapeutic drugs, nanotechnology can also be used to transfer other effective small molecules. For example, a dual delivery of MDSC-inhibiting RNAi and recruiting chemokine CCL2 has been realized by using a multilayer polymer nanocapsule in mice models of fibrosarcoma, which showed great capacity in inhibiting MDSC expansion. 182 Plebanek et al 183 identified scavenger receptor type B1, which endows a high binding affinity with spherical high-density lipoprotein, as an effective attacking target expressed on MDSC. Thus, they synthesized a high-density lipoprotein-like nanoparticle and verified its biological function in inhibiting MDSC activity and boost T-cell immune responses both in vitro and in vivo.
On the other hand, researchers are also trying to search for new specific agents for MDSC neutralization. Nagaraj et al 184 demonstrated that a member of synthetic triterpenoids, named CDDO-Me in short, has impressive utility of MDSC inhibition and immune activation in cancer. Furthermore, fraxinellone, primarily used as an anti-fibrotic drug, was found to be able to reprogram TME by increasing NK cells and T cells while decreasing regulatory B cells and MDSCs. 185 The potential therapeutic approaches to overcome MDSC-mediated resistance on ICB have been summarized in Figure 3.

Potential therapeutic approaches to overcome myeloid-derived suppressor cell (MDSC)-mediated resistance on immune-checkpoint blockade (ICB). Based on the various mechanisms used by MDSC, multiple therapeutic approaches targeting MDSC have been investigated preclinically or even entered clinical trials, including chemo-immunotherapy, molecule-targeted agent, epigenetic drugs, and nanoparticles.
Potential targets and future directions
Obviously, current therapeutic strategies and targets of MDSC still cannot meet the clinical requirements sufficiently. Therefore, explorations of potential new targets regarding MDSC regulation are underway. The immune regulatory roles played by gut microbiota in cancer have gradually been uncovered. Rutkowski et al 186 described that commensal bacteria-mediated immunosuppression through MDSC recruitment and inhibition in a TLR5-dependent manner, which subsequently promotes tumor progression. In addition, CARD9 has been identified as a protector against colon cancer through blocking commensal fungi-induced MDSC expansion at tumor site. 132 These studies provided us with evidence of mechanisms of microbiota–MDSC interactions, which bring us opportunities to open up more targets in human microbiome and related proteins for ICB efficacy enhancement. Hypoxia, which represents a hallmark of TME, has been found to promote the MDSC accumulation, 187 and hypoxia elimination is able to dramatically decrease MDSC density in prostate cancer. 188
In the future, we may not only limit our studies in MDSC-regulating network, but also broaden our horizons on the TME and even the whole immune system, especially focusing on the mutual communications between MDSC and other cells or components. Extracellular vesicles, exosomes in particular, deserve more attention for their pivotal biological information transmission function, whether they participate in MDSC regulation and how they could be used as an effective therapeutic strategy to harness ICB efficacy remains to be resolved.
Concluding Remarks
Great expectations of ICB treatment and unsolved crux of immune resistance and unstable efficacy urge us to develop novel targets to enhance ICB efficacy. Growing evidence indicates that MDSC could be an effective target to deal with immune resistance and harness ICB effects. With the gradually deepening research and understanding of the roles of MDSC in the process of tumor immune regulation, many therapeutic strategies and drugs are under explorations, and some have entered the stage of clinical trial. However, more insights and studies are required for an effective and practical MDSC-targeting therapy to emerge in the future and bring cancer patient greater survival benefits.
Footnotes
Acknowledgements
The authors give sincere appreciation to all lab members.
Author Contributions
RY and HQG designed this study. THL and TYL wrote and revised the article and contributed equally to this work. WJZ and SXX collected the related references and drew the figures. ZHZ and BFF polished the language and checked the errors. All authors have read the article and agreed the final version for publication.
Funding:
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Natural Science Foundation of China (81772727 and 81772710) and Nanjing Science and Technology Development Key Project (YKK19011).
Declaration of Conflicting Interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.