
Abstract
Cushing’s syndrome and pheochromocytomas (PCCs) are associated with endocrine hypertension. Cortisol-producing adrenal adenomas are a major cause of Cushing’s syndrome. Simultaneous occurrence of cortisol-producing adrenal adenomas and PCCs is rare. Additionally, a PCC generally produces catecholamines in proportion to its size; therefore, micro-PCCs are rarely found in clinical practice. It is unknown whether micro-PCCs produce excess catecholamines during the pre-biochemical phase. Herein, we report the case of a 53-year-old woman who was referred to our hospital for further evaluation of left adrenal incidentaloma. She had been suffering from hypertension for 7 years. Endocrine tests indicated autonomous cortisol secretion, and she was diagnosed with cortisol-producing adrenal adenoma. A laparoscopic left adrenalectomy was performed. The final pathological examination revealed an adrenocortical adenoma measuring 26 × 24 mm. In addition, a micro-PCC measuring 3 × 2 mm was incidentally found near the cortisol-secreting adrenal adenoma in the ipsilateral adrenal gland. All catecholamine biosynthetic enzymes, tyrosine hydroxylase, aromatic
Keywords
Introduction
Cushing’s syndrome comprises of symptoms and signs associated with prolonged exposure to inappropriately elevated levels of free glucocorticoids in plasma. Cortisol-secreting adrenal adenomas are responsible for approximately 10% to 15% of Cushing’s syndrome cases. 1 On the other hand, pheochromocytomas (PCCs) are tumors arising from chromaffin cells of the adrenal medulla that commonly produce catecholamines (epinephrine, norepinephrine, and dopamine). 2 Both Cushing’s syndrome and PCCs are associated with endocrine hypertension. However, simultaneous occurrence of a cortisol-secreting adenoma and PCC in the ipsilateral adrenal gland is extremely rare.
A PCC generally produces catecholamines in proportion to its size. 3 Therefore, micro-PCCs are rarely found, particularly in non-hereditary patients. Adrenal medullary nodules less than 1 cm in size were previously classified as hyperplasia. 4 However, recent molecular analyses suggest that such small nodules are, in fact, considered micro-PCCs that may exhibit malignant biological behavior.2,5,6
Serological and histological assessments of the production of adrenaline, noradrenaline, and dopamine are important for endocrinologists in predicting patient prognosis. 7 The type of catecholamine produced is associated with the degree of functional differentiation of tumor cells. PCCs that produce only norepinephrine and/or dopamine have a low degree of differentiation and are thought to exhibit higher risk of metastatic events. 8 In addition, for hereditary PCCs and paragangliomas (PPGLs), identification of the type of catecholamine produced helps predict genetic variations.9-11
It is unknown whether micro-PCCs produce excess catecholamines in the pre-biochemical phase because clinically silent micro-PCCs are rarely found in clinical practice. Herein, we report a case of a small PCC that was incidentally identified after laparoscopic adrenalectomy for cortisol-secreting adrenal adenoma. We evaluated the production of catecholamines by immunohistochemical analysis.
Case Report
A 53-year-old woman was referred to our hospital for further evaluation of a left adrenal mass that grew from 1 to 2 cm in a year. She had hypertension and dyslipidemia for 7 years and was prescribed amlodipine and pravastatin, for the respective conditions. She had no history of diabetes mellitus. She had no family history of endocrine hypertension including Cushing’s syndrome and PPGLs. In addition, she had no family history of multiple endocrine neoplasia type 2 (MEN2) or MEN2-related diseases, for instance, medullary thyroid carcinoma, primary hyperparathyroidism, and submucosal neuroma. The patient did not exhibit features of Cushing’s syndrome. Also, the patient had no spell-related symptoms or chronic symptoms associated with PCC. Her systolic blood pressure varied from 125 to 155 mmHg, and orthostatic hypotension was not observed. Her weight was 69 kg, and body mass index was 29.8 kg/m2. Magnetic resonance imaging (MRI) revealed a heterogeneous left adrenal mass with a diameter of 23 mm (Figure 1). 123I-metaiodobenzylguanidine (123I-MIBG) scintigraphy did not indicate increased uptake in the adrenal mass. Laboratory studies revealed that the serum catecholamine and urinary metanephrine levels were within the normal range. The serum adrenocorticotropic hormone (ACTH) level was relatively low, and serum cortisol levels were not suppressed after the 1 mg overnight dexamethasone suppression test (DST). In addition, after an 8 mg overnight DST, the serum cortisol level was above 1 μg/dL (Table 1). Neither plasma fractionated catecholamines nor fractionated metanephrines of a spot urine were elevated, hence the 24-hour-urinary test was not performed according to the guideline in Japan. 12 Plasma free metanephrines was not covered by health insurance in Japan at the time, so they were not available. These findings indicated a cortisol-producing adrenal adenoma; hence, a laparoscopic left adrenalectomy was performed. No hemodynamic instability was observed during anesthesia induction. Corticosteroid replacement was initiated immediately after removal of the adrenal gland. Postoperatively, her systolic blood pressure improved to 120 mmHg and stabilized.

Chemical shift magnetic resonance imaging (MRI) showing a heterogeneous adrenal mass with a diameter of 23 mm. The adrenal mass showed a mottled loss of signal intensity from the in-phase (a) to opposed-phase (b) MR images.
Laboratory studies.

Dehydroepiandrosterone sulfate.
Plasma renin activity.
Plasma aldosterone concentration.
Carcinoembryonic antigen.
The final pathology revealed an adrenocortical adenoma measuring 26 × 24 mm, in which clear and dense cells coexisted (Figure 2a, b, and d). Lipomatous changes were observed in some adenoma areas (Figure 2e). The lipomatous elements were not accompanied by hematopoietic cells, so the diagnosis of myelolipoma was abandoned. 13 The attached adrenal cortex was atrophic, consistent with the autonomous cortisol secretion by the adenoma.

(a) Positional relationship of the adrenocortical adenoma (arrow) and micro-pheochromocytoma (arrowhead). Sectional views of (b) the adrenocortical adenoma and (c) the micro-pheochromocytoma. (d) Hematoxylin-eosin (HE) staining of the adrenocortical adenoma (×200). Clear cells and dense cells coexisted. (e) Lipomatous changes were observed in some parts of the adenoma (×40). (f) HE staining of the micro-pheochromocytoma (×20). (g) HE staining of the micro-pheochromocytoma (×200). (h) Chromogranin A expression of the micro-pheochromocytoma (×200). (i) Positive reactivity for MIB-1 was found in less than 1% of the micro-pheochromocytoma (×400).
In addition to the adrenocortical adenoma, a small nodule, 3 × 2 mm in size, was incidentally found in the ipsilateral adrenal medulla (Figure 2a, c, and f). Chromogranin A-positive atypical medulla cells proliferated and were arranged in the “zellballen” pattern (Figure 2g and h). The nodule was diagnosed as micro-PCC. Cellularity was moderate (150-250 cells/U), and comedo necrosis was absent. No vascular or capsular invasion was observed. The Ki67 labeling index was <1% (Figure 2i). Immunohistochemical analyses of the catecholamine biosynthetic enzymestyrosine hydroxylase (TH; F-11, sc-25269, Santa Cruz Biotechnology, Dallas, USA), aromatic
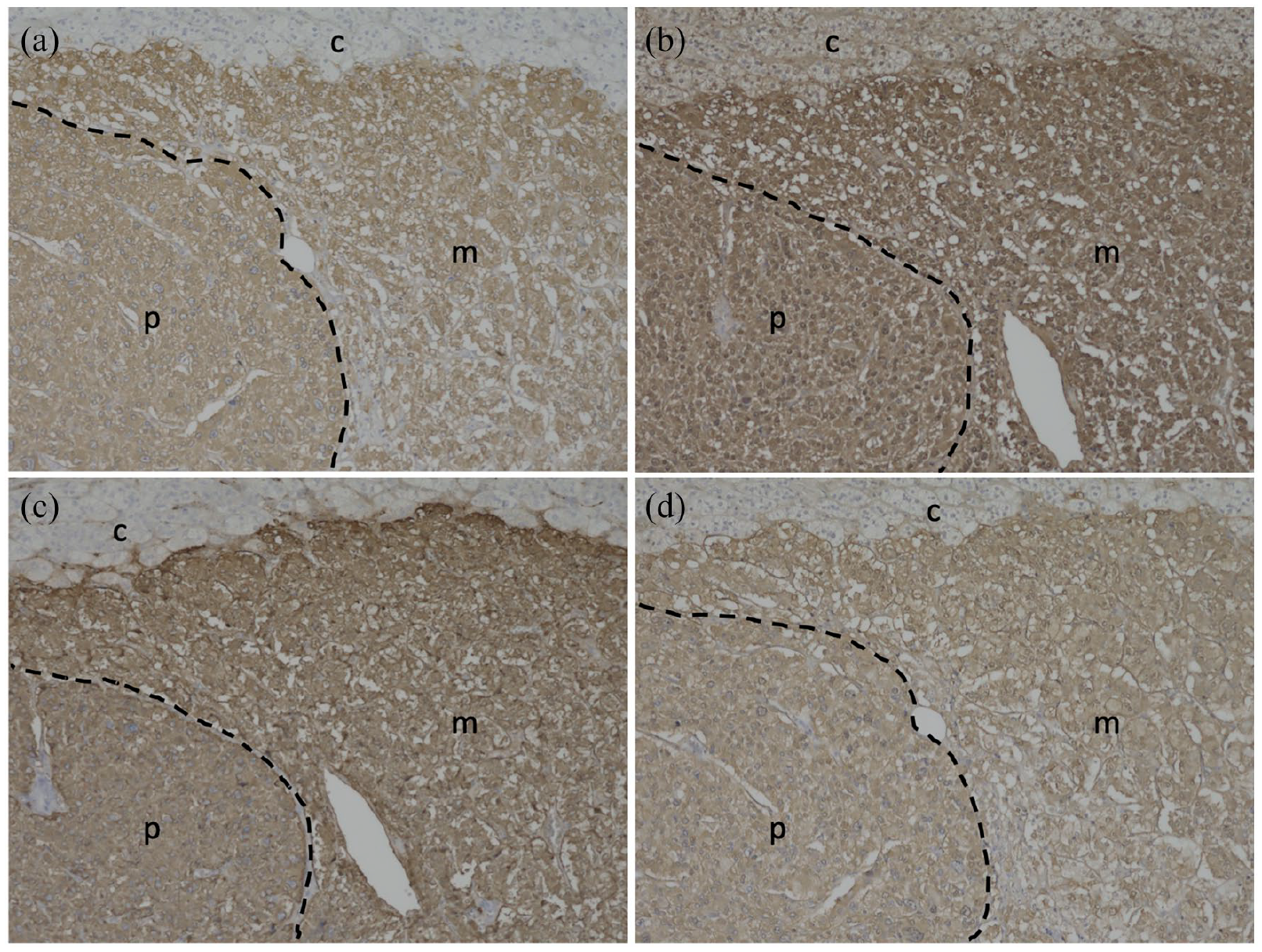
Immunohistochemical staining of the micro-pheochromocytoma with (a) tyrosine hydroxylase (TH), (b) aromatic L-amino acid decarboxylase (AADC), (c) dopamine β-hydroxylase (DBH), and (d) phenylethanolamine N-methyltransferase (PNMT) antibodies (×100). The primary antibodies were mouse monoclonal antibodies: anti-TH, F-11, sc-25269; anti-DDC, 8E8, sc-293287; and anti-PNMT, C-7, sc-393995, from Santa Cruz Biotechnology, Dallas, USA, and the anti-DBH goat polyclonal antibody, ab189991, from Abcam plc, Cambridge, UK. Dashed lines show boundaries between the normal adrenal medulla (m) and micro-pheochromocytoma (p). The letter c indicates the adrenal cortex.

Catecholamine biosynthetic enzyme expression in the normal medulla and micro-pheochromocytoma (PCC). Values are shown as percentages of the stained area. Panels (a), (b), (c), and (d) correspond to those in Figure 3.
Discussion
A micro-PCC was incidentally identified after laparoscopic adrenalectomy for cortisol-secreting adrenal adenoma. We could not predict the presence of micro-PCC based on endocrinological laboratory tests, and it was too small to be detected by imaging examinations. The levels of urinary fractionated metanephrines did not change after surgery; therefore, we could not retrospectively identify excess catecholamine productions. The micro-PCC was considered to be in the pre-biochemical phase.
In contrast, positive immunohistochemical staining for all catecholamine biosynthetic enzymes, TH, DBH, AADC, and PNMT, indicated the ability of the tumor to produce catecholamines. 14 TH, a rate-determining enzyme in the catecholamine synthetic pathway, is expressed in proportion to catecholamine production. 3 The higher expression of TH indicated that the chromaffin cells in micro-PCC autonomously produced a larger amount of catecholamines.15,16 In addition, it can be presumed that the synthesized catecholamine was epinephrine based on the positive immunostaining of PNMT, an enzyme that converts norepinephrine to epinephrine. 3
The simultaneous occurrence of a cortisol-producing adenoma and PCC distantly within the ipsilateral adrenal gland is extremely rare. To the best of our knowledge, 4 cases have been reported in the English literature (Table 2).17 -20 All patients were middle-aged, but the reason for this is unclear. Their sex, clinical presentations, and laboratory data varied.
Review of 4 cases of cortisol-producing adenoma and pheochromocytoma in the same gland (compared to the present case).
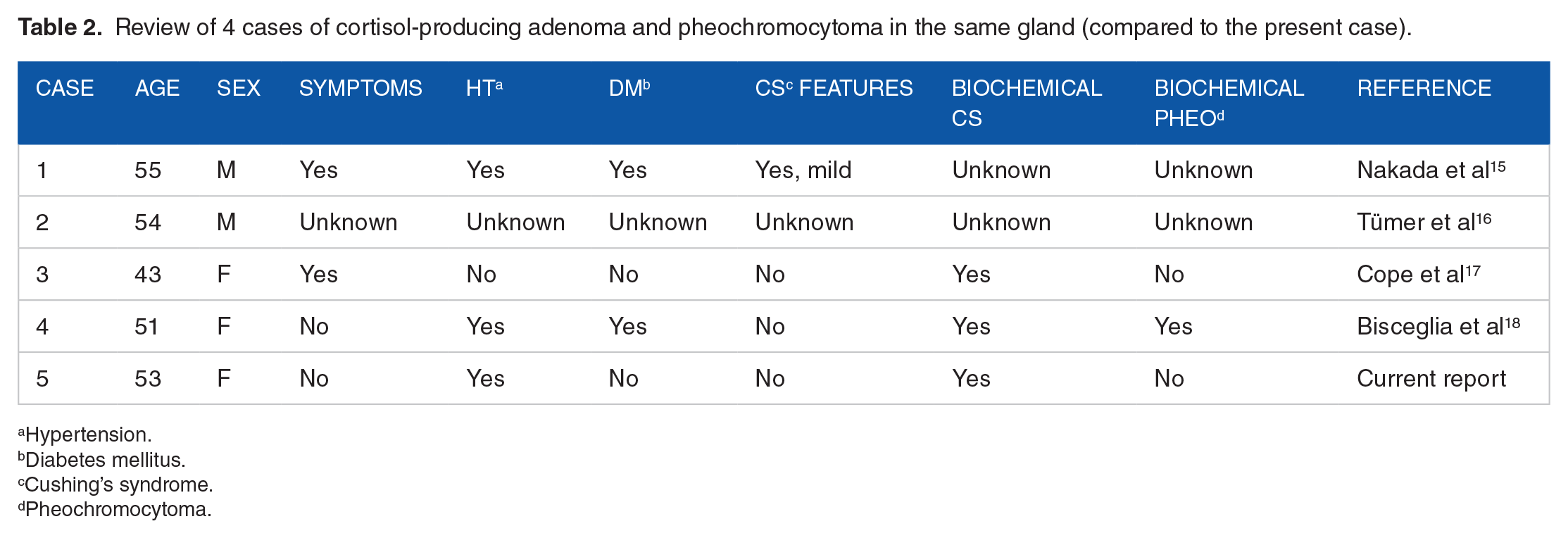
Hypertension.
Diabetes mellitus.
Cushing’s syndrome.
Pheochromocytoma.
Regarding the diagnosis of micro-PCC, the 2022 WHO classification states that adrenal medullary hyperplasia is very hard to diagnose in the condition of excess glucocorticoids. This is because adrenal medulla may look diffusely expanded due to atrophy of the adrenal cortex. 2 In our case, the lesion site in the adrenal medulla seemed to be nodular, so we considered it a micro-PCC.
Whether this patient had a hereditary PCC is a clinically important issue. Micro-PCCs that look similar to our case are commonly observed in MEN2. 2 We did not consider our case to be MEN2 because of the lack of family history and the absence of medullary thyroid carcinoma, which is usually observed from younger age with high-penetrance.
The adrenal cortex and medulla interact with each other in the local environment.21 -23 In our case, glucocorticoids secreted by the adrenocortical adenoma could have contributed to the formation of micro-PCC in the medulla. On the other hand, excess catecholamines produced by micro-PCC may have stimulated the adrenocortical adenoma and increased glucocorticoid production. Consequently, increased glucocorticoids could have contributed to the formation of lipomatous changes. We can only state that this micro-pheochromocytoma was catecholamine-producing by the fact that immunohistochemical markers are positive for catecholamine biosynthetic enzymes. If catecholamine over-secretion of this micro-PCC had been proven, we could make this hypothesis more certain. However, excess of catecholamine secretion cannot be proved because it was not detected in laboratory and imaging examinations preoperatively. Unfortunately, we could not determine catecholamine over-secretion postoperatively.
It is unclear whether micro-PCCs produce catecholamines in the pre-biochemical phase because a silent micro-PCC is rarely found in clinical practice. This is the first immunohistochemical report of a micro-PCC in the pre-biochemical phase that produced catecholamines.