
Abstract
Background:
The safe method of instructing insulin dose reduction in combination with SGLT2 inhibitors, dapagliflozin for patients with type 1 diabetes mellitus has not been clarified. In this study, we conducted a stratified, 2-arm, parallel comparative study with the primary endpoint of decreasing the frequency of hypoglycemia by instructing basal insulin dose reduction.
Methods:
The study has a multicenter, open-label, 2-arm design; 60 type 1 diabetes mellitus patients are being recruited from 7 hospitals. Study subjects have been stratified into 2 groups based on the ratio of basal insulin daily dose (Basal) to total daily insulin dose (TDD). The subjects whose Basal/TDD ratio is <0.4 are instructed not to reduce Basal but to reduce bolus insulin dose by 10% (group A), and subjects with a Basal/TDD ratio >0.4 will be instructed to reduce Basal by 10% (group B). The primary outcome is the daily frequency of hypoglycemia during the intervention period (SGLT2 inhibitor administration), as determined by self-monitoring of blood glucose. We aimed to confirm a greater reduction in frequency of hypoglycemia in group B (reduced Basal), than in group A (non-reduction of Basal and reduced insulin effect levels by 10%). Baseline hypoglycemia was set at 7 ± 6 times/month. The minimum sample size required to achieve a significance of .05 for a 1-sided t-test with a statistical power at 80% is determined. When the sample size is 26 patients in 1 group, the percentage increase in hypoglycemia exceeds 60%, and the sample size is considered sufficient.
Discussion:
In this pilot study, we assumed that, given a sufficient Basal, hypoglycemia would be more frequent in patients with type 1 diabetes when combined with SGLT2 inhibitors, provided the Basal was not reduced.
Keywords
Introduction
Background and rationale
Type 1 diabetes mellitus is a disorder characterized by absolute insulin deficiency, mainly due to autoimmune-mediated destruction of pancreatic β-cells. Although the cause of pancreatic β-cell destruction has not been completely elucidated, susceptibility genes and environmental factors have been implicated. 1 The number of patients with type 1 diabetes and absolute insulin deficiency is estimated to be approximately 100 000 to 140 000 in Japan. The age of onset is mainly from childhood to adolescence, and in Japan, the incidence rate in persons aged 0 to 19 years is 4.4/1000. 2
Because the hallmark of type 1 diabetes is absent or near-absent β-cell function, insulin treatment is essential for individuals with type 1 diabetes. 3 Insufficient provision of insulin causes not only hyperglycemia but also systematic metabolic disturbances, such as hypertriglyceridemia and ketoacidosis, as well as tissue catabolism. 3 Over the past 3 decades, evidence has accumulated supporting multiple daily injections of insulin or continuous subcutaneous administration through an insulin pump to provide the best combination of effectiveness and safety for people with type 1 diabetes. 3 Insulin therapy consists of basal insulin to maintain stable blood glucose levels and bolus insulin to control postprandial hyperglycemia and correct hyperglycemia, if necessary. The establishment of appropriate amounts of basal insulin for patients with type 1 diabetes who run out of their own insulin secretion is recommended. According to the literature, the total basal insulin dose is approximately 50% of the total daily insulin dose (TDD).4-7 Recently, Kuroda investigated the basal insulin requirement in C-peptide-negative patients with type 1 diabetes 7 and demonstrated that the basal insulin requirement is approximately 30% TDD in inpatients on diets prepared by a dietitian. 7 In addition, the maximal basal insulin requirement in all patients was 43.8% TDD, and no patients required 50% TDD. 7 Recently, King 8 also suggested that this should be revised as follows: “total daily basal insulin = 0.4 × TDD,” to prevent excess basal insulin treatment. Based on these reports,7,8 we hypothesized that the ideal basal insulin level in Asians is 30% to 40%. Based on this process, we hypothesized that a basal insulin level of 40% or higher would increase the risk of hypoglycemia during concomitant use of SGLT2 inhibitors because of excessive basal insulin levels.
Injectable and oral glucose-lowering drugs have been studied for their efficacy as adjuncts to insulin-based treatment of type 1 diabetes. 3 Therapy with only insulin may cause increased body weight, especially in patients with type 1 diabetes and excess carbohydrate intake, which increases the risk of macrovascular complications. 9 In contrast, although low-carbohydrate diets can reduce the total insulin dose and the number of self-injections, they can result in nutritional imbalances. 10 Increased mean body mass index (BMI) reported in patients with type 1 diabetes 11 also increases the risk of cardiovascular disorders. Recently, the sodium–glucose cotransporter 2 (SGLT2) inhibitor dapagliflozin, in combination with insulin therapy, was approved for the treatment of type 1 diabetes. 12 SGLT2 inhibitors reduce hyperglycemia via insulin-independent mechanisms by increasing glucose elimination via the kidneys. 12 Eight clinical trials have been published on the use of SGLT2 inhibitors as an adjunctive therapy for type 1 diabetes, in which glycated hemoglobin A1c (HbA1c), insulin dose, and body weight were decreased.13-19 The addition of an SGLT2 inhibitor to insulin therapy has been associated with improvements in HbA1C and body weight when compared with insulin alone.20-23 Thus, SGLT2 inhibitors prevent cardiovascular complications in patients with type 1 diabetes mellitus, as reported in patients with type 2 diabetes mellitus.14,24
However, SGLT2 inhibitor use is also associated with more adverse events, including hypoglycemia and ketoacidosis. There is a knowledge gap regarding the prevention of ketoacidosis or hypoglycemia risk in real-world practice. Hypoglycemia is an important determinant of glycemic control in the treatment of type 1 diabetes. 25 Many patients with type 1 diabetes struggle to achieve glycemic control and experience significant fluctuations in blood glucose levels despite insulin treatment.12,26 In randomized trials, SGLT2 inhibitors demonstrated significant reductions in HbA1c, glucose exposure, and measures of glycemic variability as well as increased time in the target glycemic range when administered as an adjunct to insulin. 12
To prevent hypoglycemia, many patients with type 1 diabetes consider reducing the basal insulin dose, which may affect glycemic control. While the addition of SGLT2 inhibitors may reduce the risk of hypoglycemia, their use has been reported to increase the frequency of ketoacidosis. 17 Therefore, reducing the insulin dose to prevent hypoglycemia during adjunctive SGLT2 inhibitor treatment may increase the risk of ketoacidosis, whereas maintaining the same insulin dose in order to prevent ketoacidosis may increase the risk of hypoglycemia. An algorithm for insulin adjustment would be beneficial when combining SGLT2 inhibitor treatment with insulin in patients with type 1 diabetes mellitus.
Objectives
We aim to explore whether the reduction of the basal insulin dose combined with SGLT2 inhibitors in patients with type 1 diabetes can reduce the frequency of hypoglycemia. We hypothesize that with an adequate basal insulin dose, the frequency of hypoglycemia will be higher if the basal insulin dose is not reduced when combined with an SGLT2 inhibitor.
Trial design
Parallel group and exploratory.
Methods: Participants, Interventions, and Outcomes
Study setting
This is a multicenter, open-label, non-randomized, exploratory, prospective, interventional study (Figure 1). The subjects are stratified into 2 groups based on the ratio of basal insulin (Basal) to the total daily insulin dose (TDD) (Basal/TDD, <0.4 or ⩾0.4). The instructions for the study subjects are shown in Table 1.
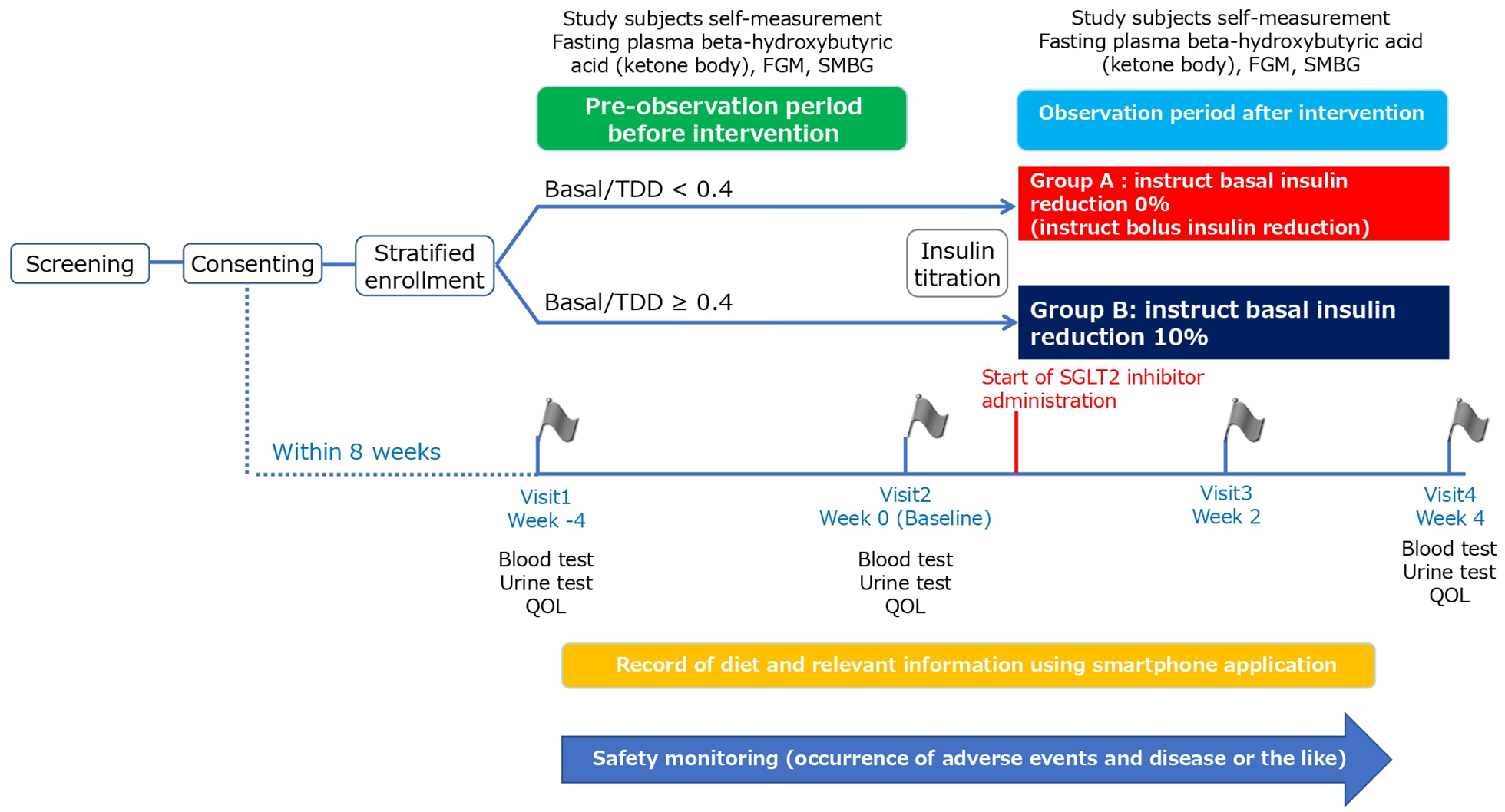
The study design. The study subjects are stratified into 2 groups based on the ratio of basal insulin (Basal) to the total daily insulin dose (TDD) (Basal/TDD, <0.4 or ⩾0.4). The study does not involve randomization of the participants.
Protocol therapy.

The diagnosis of ketoacidosis should be done at the medical consultation in the clinical institutions despite the presence/absence of subjective symptoms and plasma glucose.
Eligibility criteria
In line with the objectives of the study and to ensure the safety of the subjects, the inclusion and exclusion criteria explained in Table 2 were established. To appropriately evaluate the efficacy of the study drugs, patients who require a legal representative are excluded.
Inclusion and exclusion criteria.

Rationale for the inclusion criteria:
1 to 3: for the appropriate evaluation of the efficacy outcomes in the RISING-STAR study.
4: for the safety of the study subjects in a real-world situation.
5: for the free-will participation of the study subjects.

Who will take informed consent?
In general, informed consent form is used to obtain participants’ consent before they join the study. Investigators use informed consent form approved by the certified review board to provide adequate explanations to patients and confirm their full understanding of the contents provided before obtaining their written consent. Only patients who are able to give their consent in writing are included in this study. Those patients who need their legal representative’s consent is not allowed to participate.
Additional consent provisions for collection and use of participant data and biological specimens
Not applicable.
Interventions
Explanation for the choice of comparators
The randomized assignment is not conducted in the study. Basal/TDD at giving their consent is used to stratify the study subjects to the Group A or the Group B.
[Group A] Study subjects reduce the basal insulin dose by 0% (instructed to reduce bolus insulin dose).
Subjects whose Basal/TDD ratio is <0.4 are instructed not to reduce basal insulin dose but to reduce bolus insulin dose by 10%. The 10% reduction of the bolus insulin dose is based on a 90% carbohydrate insulin ratio (CIR), and it is rounded to the nearest whole number where insulin therapy involves multiple daily injections (MDI) or rounded down to 2 decimal places in cases of continuous subcutaneous insulin infusion (CSII). The study subjects are instructed to follow this instruction for 3 days from the start of the intervention. After the fourth day, subjects can titrate both the basal and bolus dose according to the “Algorithm for Basal Insulin Titration after SGLT2 administration (Figure 2)” and “Algorithm for Bolus Insulin Titration after SGLT2 administration (Figure 3).”

Algorism for basal insulin titration after SGLT2 administration. The study subjects are to follow the dosing instruction for 3 days from the start of the intervention, after which the basal and bolus insulin can be titrated either by the subject or by instruction from the attending physician according to “algorithm for basal insulin titration after SGLT2 administration.”
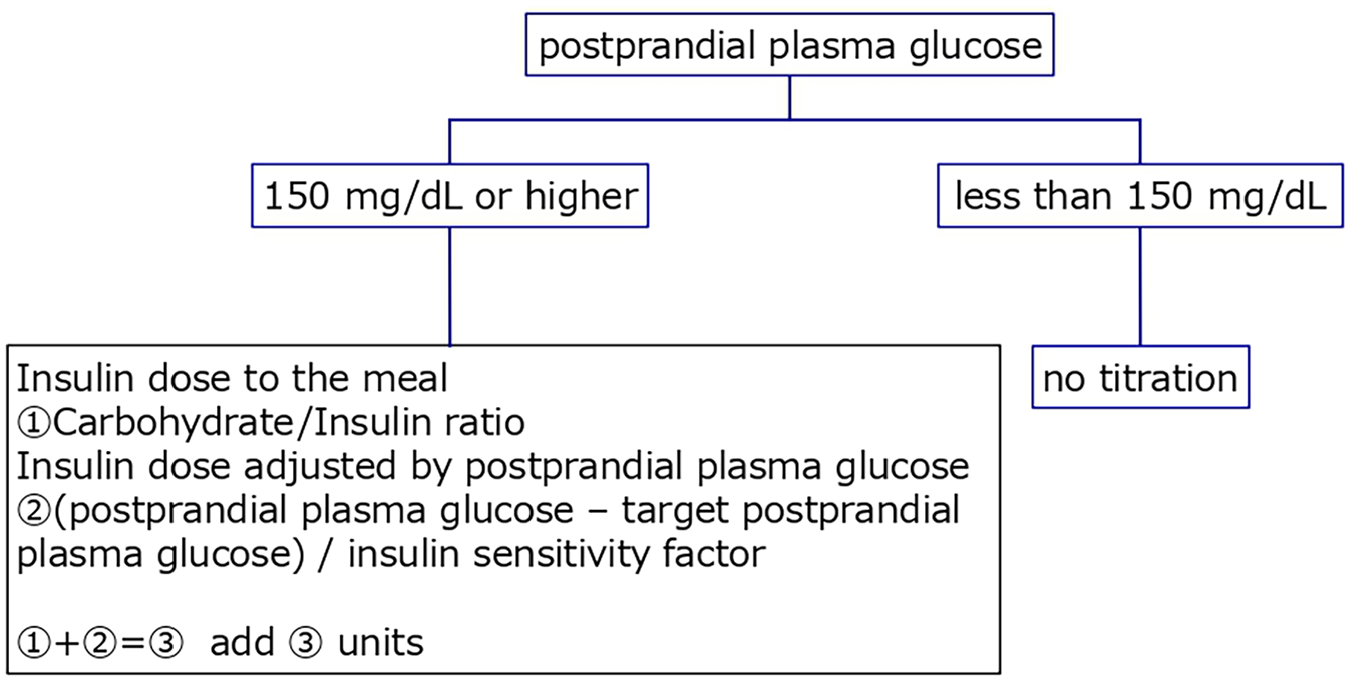
Algorism for Bolus insulin titration after SGLT2 administration. The study subjects are to follow the dosing instruction for 3 days from the start of the intervention, after which the basal and bolus insulin could be titrated either by the subject or by instruction from the attending physician according to “algorithm for Bolus insulin titration after SGLT2 administration.”
[Group B] Study subjects are instructed to reduce basal insulin by 10%.
Study subjects whose Basal/TDD is ⩾0.4 are instructed to reduce the total insulin dose by 10%, reducing the basal insulin dose only. The dose of basal insulin is rounded down to 1 decimal place in cases of MDI or to 2 decimal places in cases of CSII. The study subjects are instructed to follow this instruction for 3 days from the start of the intervention. After the fourth day, subjects can titrate both the basal and bolus dose according to the “Algorithm for Basal Insulin Titration after SGLT2 administration (Figure 2)” and “Algorithm for Bolus Insulin Titration after SGLT2 administration (Figure 3).”
Intervention description
A pre-observation period of 4 weeks is established before the intervention (administration of SGLT2 inhibitor). During this period, study subjects will measure and record fasting plasma beta-hydroxybutyric acid (BOHB) levels, flash glucose monitoring (FGM), and self-monitoring of blood glucose (SMBG). The initiation of SGLT2 inhibitor administration is set as day 0 of the observation period. The study subjects will visit the research institutions 4 times: at weeks −4, 0, 2, and 4. The study subjects are provided with a digital camera and instructed to take a picture of each meal they eat throughout the observation period.
Criteria for discontinuing or modifying allocated interventions
Criteria and coping for discontinuation of the study
If, for the reasons listed below, the investigators determine that a subject can no longer continue to participate in the study, necessary measures such as discontinuing administration of the study agent, adjustment of insulin type and dose, etc. will be immediately taken. Data on the subject will be handled as data of a “study discontinuation case.” The date, time point in the study, reason for discontinuation, and the subject’s course of the disease will be entered onto the carte and the CRF. Necessary tests will also be performed upon discontinuation. The efficacy and safety of the subject`s treatment will be evaluated at this point. Moreover, even if the subject’s participation is discontinued, items to be evaluated will be followed up as much as possible for the purpose of analysing safety.
Criteria for discontinuation of study in each study subject
1) When Investigators judge the discontinuation is necessary at sick day or physical deconditioning.
2) When the investigators diagnose as diabetic ketoacidosis.
3) When a study subject voluntarily wants to discontinue the study or withdraw her/his consent.
4) When a major nonconformance is found not after the registration.
5) When continuation of using the study agent is not appropriate due to worsened primary disease or complications.
6) When discontinuation of the study is necessary due to occurrence of adverse events and disease or the like.
7) When patients are found pregnant.
8) In case of remarkable poor adherence (medication rate is expected to less than 75% or 120% or higher to the planned administration).
9) When investigators judge discontinuation of the study is appropriate due to other reasons.
Criteria for discontinuation of study
1) When the study is difficult to continue due to any of the following reasons, the principal investigator decides whether the study should continue. If continuation is determined to be inappropriate, the principal investigator shall inform the decision of the discontinuation to the responsible investigators of all the collaborating institutions as soon as possible along with the underlying reasons, and information of how to respond to their participants in order to take necessary measures. The principal investigators shall report the discontinuation of the study to the certified review board by written document.
1-1) When critical information about quality, safety, and efficacy of the study agent is obtained.
1-2) When recruiting of study subjects is difficult and the planed number of study subject to be enrolled is determined difficult to achieve.
1-3) When the objective of the study is accomplished before reaching the planned number of enrollment or the planned period.
1-4) When protocol modification is instructed but unable to execute.
2) When discontinuation of the study takes place, the responsible investigators promptly discontinue the study and make a report to the head of their institution by written document. Investigators also need to notify the discontinuation of study to their participants and take other appropriate actions.
Deviation from the protocol
When investigators need to deviate from or modify the study protocol in order to avoid an emergency risk of their study subjects or for other medically compelling reasons, the content and reason shall be stated in the carte and the CRF. Investigators also follow such study subjects as much as possible during the study. Even if investigators deviate from the protocol, they need to continue collecting necessary information as much as possible for this study. For data handling, it is decided by the data handling committees under a blinded circumstance.
Management of incompatibility
Incompatibility is defined as noncompliance with legal regulations, protocol and operation procedures, and falsification or fabrication of study data.
The incompatibility will be managed as follows.
When responsible investigator recognizes that the incompatibility of the study, the responsible investigator promptly reports the fact to the head of the institution and the principal investigator.
When investigator recognizes that the incompatibility of the study, the investigator promptly reports the fact to the responsible investigator.
The principal investigator promptly hears the opinion from the certified review board in case of the specifically serious incompatibility.
Strategies to improve adherence to interventions
Management of the study agent
This study does not use placebo. Both groups use approved, commercially available drug as the study agent. Also, this study is open-label study. Therefore, specific management of the study agent is not conducted in the study, and the study agent is managed as same as general drugs.
Relevant concomitant care permitted or prohibited during the trial
Combination therapy, restricted concomitant drugs, and prohibited concomitant drugs.
The study subjects receive by intensive-insulin therapy or insulin-pump therapy as basal treatment other than dapagliflozin as therapeutic intervention. Type of insulin is not restricted, but the change of usage or dose is conducted according to insulin titration algorism described in Figure 2. Algorism for Basal Insulin Titration after SGLT2 administration and Figure 3. Algorism for Bolus Insulin Titration after SGLT2 administration.
The concomitant anti-diabetic agents other than insulin is not restricted, but in principal addition of new medication, withdrawal or change of medication, change in usage or dose of medication is not conducted during the observation period in both groups for appropriate evaluation.
Provisions for post-trial care
This study is conducted within the scope of approval treatment with the approved drug which is commonly performed. Therefore, generally, special compensation is not provided even when health damages are caused by the study drug. Handling the incidence is exactly same as a health damage or medical accident that occurs while a patient receives a regular medical attention. The compensation of such case is based on the Adverse Drug Reaction Relief System of the Pharmaceutical and Medical Devices Agency, Product Liability Law, or product liability insurance.
In case, health damage is occurred other than medical practice, the principal investigator has to buy the insurance for clinical study on behalf of other investigators since the design of this study is interventional.
Outcomes
Observations and schedules are shown in Table 3. In principle, the study subjects will visit the research institutions, and at every visit, blood tests (fasting) and urine tests (spot) are performed. Investigators collect and enter the results of the examinations listed in Table 3 in the case report form (CRF) and send the CRF to the data center. Adverse events are followed as safety endpoints throughout the study. The items measured by the study subjects themselves are recorded in specific documents and sent to the data center via the investigators.
Observation items.

Obtained by calculation.
Observation is conducted before the start of study agent administration.
Participant timeline
Observations and schedules are shown in Table 4.
Observation schedule.

○ Item is required to be observed at indicated observation period/observation point.
△Item is optionally observed at indicated observation period/observation point.
▲Item is observed if a residual sample exists.
Observation is conducted before the start of study agent administration.
Sample size
The RISING-STAR study is designed as an exploratory study, and no prior studies have reported the frequency of hypoglycemia after the administration of SGLT2 inhibitors. We hypothesized that the frequency of hypoglycemia per day would increase after the administration of an SGLT2 inhibitor, provided the dose of basal insulin was not titrated. The baseline frequency of hypoglycemia was set at 7 ± 6 times/month when insulin glargine was used in patients with type 1 diabetes. 29 The minimum sample size required to achieve a significance of .05 for a 1-sided t-test with a statistical power at 80% is determined. When the sample size is 26 patients in 1 group, the percentage increase in hypoglycemia is more than 60%, and the sample size is considered sufficient (Figure 4). With an estimated dropout rate of 10%, the planned number of subjects (60 subjects, with 30 in each group) is considered to have adequate statistical power for an increased hypoglycemia frequency of more than 60% (11.2 ± 6 times/month) in Group A (Basal/TDD <0.4, with study subjects instructed to reduce basal insulin dose by 0% and bolus insulin dose by 10%) from 7 ± 6 times/month in Group B (Basal/TDD ⩾0.4, with study subjects instructed to reduce basal insulin by 10%).
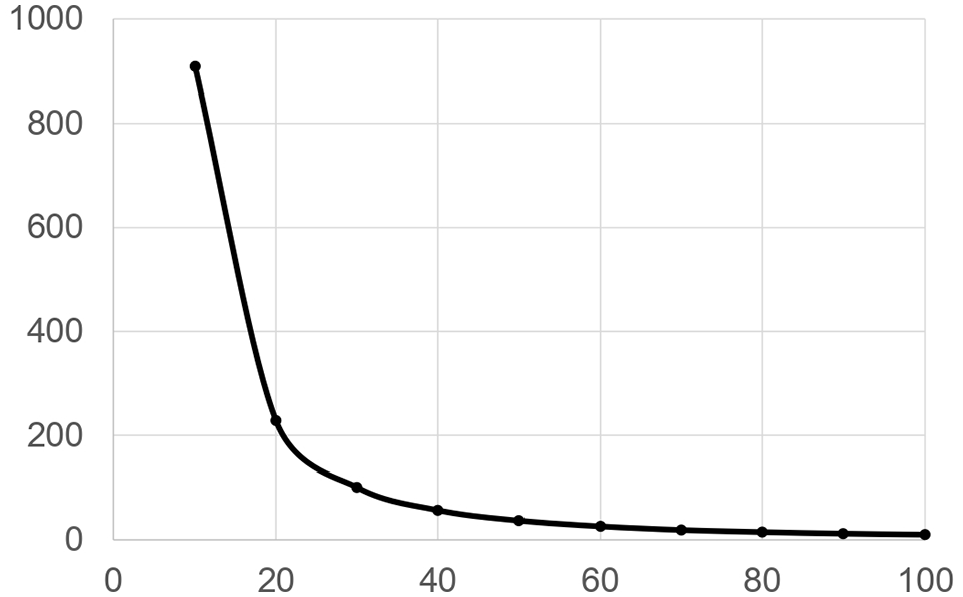
The assumed increase of hypoglycemia and required sample size. Baseline hypoglycemia was set at 7 ± 6 times/month. An increase in hypoglycemia is expressed as a percentage increase from the baseline (shown on the X-axis). The minimum sample size required to achieve a significance of .05 for a 1-sided t-test with a statistical power at 80% is determined. The necessary sample size is expressed as the number of patients in 1 arm (shown on the Y-axis). When 26 patients in 1 group were analyzed, the percentage increase in hypoglycemia was more than 60%, and the sample size was estimated to be sufficient.
The RISING-STAR study was conducted at 7 research institutions where a total of 350 patients with type 1 diabetes are being treated. According to a previous survey, 30% of these patients were eligible for SGLT2 inhibitor treatment, as per Japanese labeled indications, meaning that approximately 100 patients could potentially use the SGLT2 inhibitor with insulin. Among the 100 patients, all patients who sign a written consent form are enrolled in the study and stratified into Group A (Basal/TDD <0.4, subjects instructed to reduce basal insulin dose by 0% and bolus insulin dose by 10%) or Group B (Basal/TDD ⩾0.4, subjects instructed to reduce basal insulin by 10%). The proportion of patients who will consent is assumed to be 80%, and the proportion of patients who meet the inclusion criteria and none of the exclusion criteria is assumed to be 80%. Under these conditions, a target number of 60 study subjects is feasible.
Recruitment
This study is conducted in 7 research institutions, and total 350 patients with type 1 diabetes mellitus is under treatment in these institutions. From the previous survey, 30% of these patients are assumed to be eligible for the use of SGLT2 inhibitor, meaning that approximately 100 patients can use SGLT2 inhibitor with insulin. Among the 100 patients, all patients who provide written consent form within enrollment period are entered into the study and are stratified into the Group A or the Group B based on Basal/TDD. Proportion of patients who give consent is assumed to 80%, and proportion of patients who fall into any of the exclusion criteria is assumed to 80%.
Assignment of Interventions: Allocation
Sequence generation
Not applicable.
Concealment mechanism
Not applicable.
Implementation
Not applicable.
Assignment of Interventions: Blinding
Who will be blinded
Not applicable.
Procedure for unblinding if needed
Not applicable.
Data Collection and Management
Plans for assessment and collection of outcomes
Source documents
The research institutions store and manage the following information as the source documents, and respond to the request from monitoring, audit or the certified review board.
Source documents for each data (medical records, nursing records, test data, medication records, CRF, study subjects’ diary, QOL questionnaire, etc.).
Informed consent records showing that the subjects consent to participate in this study.
Documents to be considered as source documents
The research institutions store and manage the following information as documents to be considered as the source documents in addition to the source documents, and respond to the request from monitoring, audit, or the certified review board.
consent withdrawal form.
information of medication adherence.
information of adverse events and disease or the like.
Plans to promote participant retention and complete follow-up
Handling of adverse event
During the study period, the occurrence of any untoward medical events experienced by study subjects, including the worsening of a pre-existing underlying disease, will be defined as adverse events. If complications worsen after starting the study, this will also be defined as an adverse event. However, the worsening of efficacy endpoints within the reference values in each research institution will not be treated as an adverse event.
Coping with adverse event
If a study subject experiences an adverse event during the study period, the investigators will immediately take the appropriate medical action. In accordance with the procedures of each participating research institution, they will also generate a report to the responsible investigator and the head of the institution and enter the necessary information onto the carte and the CRF. If suspension of the study drug administration or medical treatment for the adverse event is needed, the study subject has to be informed. Regarding the handling of a serious adverse event.
Data management
Central registration numbers are used to identify the study subjects. When transferring data of the study subjects in a form of electronic data, approval of the person responsible for the data management is required. When transferring data from an electronic network that is not secured, the data must be encrypted at the sender site. When data center needs to provide data of the study subjects to the outside, approval of the principal investigator, and the person responsible for the data management is required. In case of the study subjects who discontinued the study, tests are conducted as much as possible, and enter the data till the study discontinuation to the database.
Confidentiality
Central registration numbers are used to identify the study subjects. When transferring data of the study subjects in a form of electronic data, approval of the person responsible for the data management is required. When transferring data from an electronic network that is not secured, the data must be encrypted at the sender site. When data center needs to provide data of the study subjects to the outside, approval of the principal investigator, and the person responsible for the data management is required. In case of the study subjects who discontinued the study, tests are conducted as much as possible, and enter the data till the study discontinuation to the database.
Plans for collection, laboratory evaluation and storage of biological specimens for genetic or molecular analysis in this trial/future use
All parties involved in this study must make their best efforts to protect personal information of study subjects. This study is conducted in accordance with Personal Information Protection Law, and other legislation/law and regulation. Unique information (initial, carte number) of study subjects is stored securely in the research institutions, and information that allows a person outside the research institution to identify the study subjects (such as name, address, telephone number, etc.) is not included in CRFs and registration database.
A central registration number is used when the data center inquires for the data of the study subject to the research institution. Investigators use the correspondence tables to identify their research subjects (anonymization), which are managed by themselves. Investigators store the correspondence tables securely and keep them properly during the storage period prescribed in the Clinical Trial Act (until the day after a lapse of 5 years from the day on which the completion of the study), or the storage period prescribed at each research institution, whichever comes later. The person responsible for personal information management at each research institution shall be the responsible investigator unless otherwise specified.
The investigators, and the heads of the research institutions they belong to store the study-related information properly during the storage period prescribed in the Clinical Trial Act (until the day after a lapse of 5 years from the day on which the completion of the study), or the storage period prescribed at each research institution, whichever comes later. The responsible person in charge of data management also stores the study-related information such as CRF as the source documents and electronic information such as data sets properly in same way.
After the storage period, the data must be destroyed not to be restored by the data carrier.
Anonymized data collected for the analysis is kept stored for future secondary study such as meta-analysis. If the anonymized data is used for other studies, approval from the ethics review board is required before the study implementation.
Samples for special blood tests are measured in laboratory companies utilized in each research institution, and after obtaining data, they are disposed under the responsibilities and procedures of the companies. The companies do not store the sample.
Statistical methods
Statistical methods for primary and secondary outcomes
Analysis of the primary endpoint
Summary statistics will be calculated for the number of hypoglycemic events per day (plasma glucose level defined as SMBG ⩽70 mg/dL) during the intervention period (from baseline to week 4) using the full analysis set (FAS) as the main analysis set and the per protocol analysis set as the sensibility analysis set. For comparisons between groups, a 2-sample t-test and analysis of covariance will be conducted and differences between the groups as well as their 95% confidence intervals calculated. HbA1c, age, and frequency of hypoglycemia (⩽70 mg/dL, confirmed by SMBG) per day during the pre-observation period before the intervention (week −4 to baseline) will be used as covariates in the analysis of covariance. If the data distribution does not follow a normal distribution, the summary statistics will be calculated after logarithmic transformation.
Analysis of the secondary endpoints
For the analysis of secondary endpoint 1 (frequency of ketosis) and 2 (frequency of hypoglycemia detected by FGM), summary statistics of measurements and changes will be calculated using FAS during the pre-observation period before the intervention (week −4 to baseline) and the observation period after the intervention (baseline to week 4) in each group. For the measurements, a 2-sample t-test will be used for comparisons between groups, and for the change, a 1-sample t-test will be used for comparison in each group. If the data distribution does not follow a normal distribution, the summary statistics will be calculated after logarithmic transformation.
For the analysis of secondary endpoint 3, summary statistics will be calculated using FAS for measurements at each observation point and change in the measurements from baseline to each observation point after the intervention. For the measurement, a 2-sample t-test will be used for comparisons between groups, and for the change in the measurement, a 1-sample t-test will be used for comparison in each group.
Analysis of the safety endpoints
To analyze the safety endpoints, a table of all adverse events and diseases will be created for each group using the safety analysis set, and comparisons will be performed between groups as necessary using Fisher’s exact test.
Interim analyses
Not applicable.
Methods for additional analyses (eg, subgroup analyses)
To analyze the correlation of change in dietary habit, dietary content, and nutrient intake with change in fasting plasma BOHB acid, Spearman rank correlation coefficient, and its 95% confidence interval will be calculated and evaluated for significance. The subjects are instructed to take pictures of each meal using a digital camera. The stored images will be uploaded to a cloud, and diabetologists will analyze the images according to the Standard Tables of Food Composition in Japan using a specialized application (Asken Wit Co. Ltd.). The volumes and calories of carbohydrates, proteins, fats, and nutrient intakes will be calculated using the system referred to as “online nutritional evaluation.”
Methods in analysis to handle protocol non-adherence and any statistical methods to handle missing data
The data relating Protocol non-adherence is not included in the per protocol population but is included in full analysis. Imputation is not planned to use for missing data.
Plans to give access to the full protocol, participant level-data, and statistical code
This study has no plan for granting public access to the full protocol, participant-level dataset, and statistical code.
Oversight and Monitoring
Composition of the coordinating centre and trial steering committee
Data Center (Data Management)
Yoshifumi Sezutsu
Data management group, Clinical Research Support Division, Soiken Inc.
Location: Nikko Building 5F, 1-1 Kanda-Ogawamachi, Chiyoda-ku, Tokyo
Tel: 03-3518-9918
Data Center conducts data management works in the study according to the operation procedure.
Monitoring
Yasunori Tomita
Monitoring group, Clinical Research Support Division, Soiken Inc.
Location: NBF Building 4F, 1-3-1, Kanda-Ogawamachi, Chiyoda-ku, Tokyo
Tel: 03-3295-1350
The person in charge of monitoring conducts monitoring works in the study according to the operation procedure.
Audit
Toru Matsumura
Quality Assurance group, Clinical Research Support Division, Soiken Inc.
Location: NBF Building 4F, 1-3-1, Kanda-Ogawamachi, Chiyoda-ku, Tokyo
Tel: 03-3295-1350
The person in charge of audit conducts audit in the study according to the audit plan and the operation procedure, and assesses whether the data quality management is appropriately conducted in the study.
Data Handling Committee
The data handling committee decides all the data handling including missing data and data deviated from the protocol, under a blinded circumstance before statistical analyses. The committee consists of the principal investigator, the responsible officer, and the biomedical statistics expert.
Composition of the data monitoring committee, its role and reporting structure
Monitoring performed in accordance of the standard operation procedure of the monitoring by a third-party organization, Soiken Inc. Regarding the quality of data, the principal investigator investigate the progress of the study through monitoring personnel periodically and the research conducted in compliance with the study protocol and the Ethical Guidelines for Medical and the Clinical Trial Act, and take appropriate measures to prevent any deviation from the protocol. In addition, the monitoring personnel make a monitoring report and submit it periodically to the principal investigator. To ensure the safety of the study subjects, adverse events and the diseases or the like are monitored and promptly reported.
Adverse event reporting and harms
Serious adverse event (SAE)
A serious adverse event refers to, regardless of the presence or absence of a causal relationship with the study agent, a sign, symptom, or disease undesirable or unintended for the study subject that meets the “Ethical Guidelines for Medical and Health Research Involving Human Subjects (Chapter 1, Part 2).”
During this study, when serious adverse event occurs, regardless of the presence or absence of a causal relationship with this study, investigators immediately conduct appropriate treatment for the study subject.
[Serious adverse events]
Those that lead to death.
Those are life-threatening.
Those that require hospitalization or extension of hospitalization for treatment.
Those that result in permanent or significant disability or malfunction.
Other events or reactions medically important or critical.
Those are equivalently severe to 1 to 5.
Those that could be transmitted to descendants.
Reporting of adverse event
1. Immediate reporting
When an adverse event subject to immediate reporting (a serious adverse event [SAE]) occurs during this study, an investigator promptly explains the occurrence to the study subject takes appropriate treatment. Also, regardless of the presence or absence of a causal relationship with the study agent, the investigator reports it appropriately to the data center within 5 days, and to the authorized head of the research institution in accordance with “the Ethical Guidelines for Medical and Health Research Involving Human Subjects (partially revised on February 28, 2017),” the Clinical Trial Act, and procedures of the research institution. The data center compiles the information, takes direction of the principal investigator, and reports the fact to the responsible investigators of the collaborating research institutions. In addition, the information needs to be notified to the company manufacturing/selling the study agent (AstraZeneca K.K. and Ono Pharmaceutical Co. Ltd.) by the SAE/disease or the like Reporting Form including the name of the research institution, the name of attending physician, causality of the events in relation to all study medication, and the ESR number of the study (ESR-19-14385).
2. Regular reporting
When an adverse event other than SAE occurs, describe it in the CRF corresponding to the time of occurrence, and report it to the data center by sending the CRF. The data center compiles the information appropriately, and reports it to the principal investigator to obtain directions on how to respond. In addition, the information needs to be notified to the company manufacturing/selling the study agent (AstraZeneca K.K. and Ono Pharmaceutical Co. Ltd.).
Frequency and plans for auditing trial conduct
Audit is performed by a third-party organization, Soiken Inc. An auditor performs it in accordance with the protocol and the standard operation procedure to checks whether or not this study has followed the study protocol appropriately as stated. The results shall be reported by the auditor to the responsible investigator, principal investigator, and the heads of the participating institutions.
Plans for communicating important protocol amendments to relevant parties (eg, trial participants, ethical committees)
When protocol is modified, the modification is reported to Kyoto Prefectural University of Medicine Clinical Research Review Board immediately. After protocol modifications is approved, the modification is reported to jRCT.
Dissemination plans
The trial results will be published at peer-reviewed international journals.
Discussion
The findings will provide knowledge regarding the reduction of the basal insulin dose combined with an SGLT2 inhibitor in patients with type 1 diabetes, which may reduce the frequency of hypoglycemia associated with the combination therapy. The results will be disseminated through presentations at appropriate conferences, meetings, and publications in peer-reviewed journals.
In this pilot study, we assumed that, given sufficient basal insulin doses, hypoglycemia would be more frequent in patients with type 1 diabetes when combined with SGLT2 inhibitors if basal insulin doses were not reduced. The best study design to clarify this question is to observe hypoglycemic attacks as the primary endpoint in type 1 diabetes with SGLT2 inhibitors or placebo. In this study, the best study design would be to stratify participants according to the ratio of basal insulin to total insulin and to determine the association between basal insulin and total insulin as well as hypoglycemic attacks. However, we assumed from previous reports that a 10% reduction in total insulin dose is necessary to avoid hypoglycemia with SGLT2 inhibitor treatment.12,15,16
SGLT2 inhibitor treatment in patients with type 1 diabetes mellitus has been reported to improve glycemic control. 12 The amount of additional insulin is determined from carbohydrate and preprandial blood glucose intake and treatment target blood glucose levels using applied carb counting in type 1 diabetes patients covered by this pilot study. In this pilot study, we envisioned avoiding hypoglycemia in SGLT2 inhibitor therapy by reducing the basal insulin dose, the insulin effect value, or both. In this pilot study, after adhering to their doctor’s insulin reduction instructions for 3 days, patients will be allowed to adjust their own insulin dose appropriately by watching their blood glucose levels over those 3 days. Furthermore, subjects will be divided into 2 groups: a 10% reduction in basal insulin dose or a 10% reduction in insulin effect dose. In addition, categorization into the 2 groups will not be randomized but will instead utilize insulin levels prior to SGLT2 inhibitor treatment, with a 10% reduction in basal insulin in patients with basal insulin levels greater than 40% of total insulin and a 10% reduction in insulin effect size in patients with basal insulin levels less than 40% of total insulin We assumed that the risk of developing hypoglycemia would be increased in the group that did not indicate insulin reduction, and we did not establish a group that did not indicate insulin reduction.
We assumed that when insulin therapy was combined with an SGLT2 inhibitor for patients with type 1 diabetes mellitus, the frequency of hypoglycemia would increase if the basal insulin dose was not reduced, provided there was an adequate basal insulin dose. In cases where the basal insulin dose was less than 40% of the total insulin dose, there was a possibility that an inadequate basal insulin dose was not administered and that the amount of additional insulin was excessive. The cause of the overdose of additional insulin may be that the amount of carbohydrate ingested is excessive and that the insulin effect value is estimated to be higher than the true value. To account for these possibilities, in cases where basal insulin levels were less than 40% of total insulin, we did not indicate a reduction in basal insulin levels but rather a 10% reduction in insulin effect values.
The purpose of this pilot study was to analyze the feasibility of future randomized trials. The first randomized trial was envisioned as a 2-arm, randomized trial in which a reduction in basal insulin dose was indicated or no reduction in basal insulin dose was instructed for concomitant use of SGLT2 inhibitors in patients with type 1 diabetes in order to prevent the development of hypoglycemia. In order to conduct this randomized trial, we need to determine the risk of diabetic ketoacidosis with SGLT2 inhibitor treatment in type 1 diabetes; SGLT2 inhibitor treatment in type 1 diabetes may increase the risk of diabetic ketoacidosis 6-fold.20,30 The U.S. Food and Drug Administration estimates that 1 additional case of ketoacidosis occurs every 26 years when patients with type 1 diabetes are treated with sotagliflozin. Assuming a case fatality rate of 0% to 4%, this estimate corresponds to 16 additional deaths per year per 100 000 patients with type 1 diabetes.31,32 Thus, although a reduction in basal insulin dose in SGLT2 inhibitor therapy in type 1 diabetes is beneficial in the prevention of hypoglycemia, there is a risk of increasing the risk of diabetic ketoacidosis. 17 In this pilot study, prevention of hypoglycemic attacks was the primary endpoint, with home self-ketone measurements as a secondary endpoint, which provides insight into the increase in ketones in response to a reduction in basal insulin dose. This will be essential for the design of future randomized trials.
The first clinical question is whether reducing the basal insulin dose to prevent the development of hypoglycemia is a risk factor for diabetic ketoacidosis in SGLT2 inhibitor therapy in type 1 diabetes. In addition, does SGLT2 inhibitor treatment increase the risk of diabetic ketoacidosis in type 1 diabetes without reducing basal insulin levels? To clarify the purpose of this study, the frequency of ketosis may also be the primary endpoint of this pilot study. However, in this pilot study, for safety reasons, the attending physician will intervene if the home autologous ketone body measurement is greater than 600 μM.27,28 This will allow the attending physician to intervene when ketones are increased and, thus, prevent ketoacidosis. Therefore, it is not possible to measure the frequency of ketosis onset in the absence of bias. Therefore, the frequency of ketosis onset was not the primary endpoint.
A second future randomized trial, with the development of ketoacidosis as the primary endpoint, will compare the basal insulin dose reduction of up to 10% in this trial with a basal insulin dose reduction of more than 10% in this trial for patients with type 1 diabetes, assuming that it is. It is also important to know whether ketoacidosis occurs in this study, in which the basal insulin dose is reduced by up to 10%.
Footnotes
Acknowledgements
Funding:
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The RISING-STAR study, including the article processing charge, is funded by AstraZeneca K.K. and Ono Pharmaceutical Co., Ltd. No drugs will be donated or funded by the sponsor. The funding bodies had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Declaration of conflicting interests:
M. H. received grants from Asahi Kasei Pharma, Nippon Boehringer Ingelheim Co. Ltd., Mitsubishi Tanabe Pharma Corp., Daiichi Sankyo Co. Ltd., Sanofi K.K., Takeda Pharma Co. Ltd., Astellas Pharma Inc., Kyowa Kirin Co. Ltd., Sumitomo Dainippon Pharma Co. Ltd., Novo Nordisk Pharma Ltd., and Eli Lilly Japan KK, outside the submitted work. M. A. received personal fees from Novo Nordisk Pharma Ltd., Abbott Japan Co. Ltd., AstraZeneca plc, Kowa Pharma Co. Ltd., Ono Pharma Co. Ltd., Sumitomo Dainippon Pharma Co. Ltd., and Takeda Pharma Co. Ltd., outside the submitted work. M. Y. received personal fees from MSD K.K., Sumitomo Dainippon Pharma Co. Ltd., Kowa Pharma Co. Ltd., AstraZeneca plc., Takeda Pharma Co. Ltd, Kyowa Kirin Co. Ltd., Daiichi Sankyo Co. Ltd., Kowa Pharma Co. Ltd., and Ono Pharma Co. Ltd., outside the submitted work. Y. H. received grants from Asahi Kasei Pharma and personal fees from Daiichi Sankyo Co. Ltd., Mitsubishi Tanabe Pharma Corp., Sanofi K.K., and Novo Nordisk Pharma Ltd., outside the submitted work. M. F. received grants from Nippon Boehringer Ingelheim Co. Ltd., Kissei Pharma Co. Ltd., Mitsubishi Tanabe Pharma Corp., Daiichi Sankyo Co. Ltd., Sanofi K.K., Takeda Pharma Co. Ltd., Astellas Pharma Inc., MSD K.K., Kyowa Kirin Co., Ltd., Sumitomo Dainippon Pharma Co., Ltd., Kowa Pharma Co. Ltd., Novo Nordisk Pharma Ltd., Ono Pharma Co. Ltd., Sanwa Kagaku Kenkyusho Co. Ltd., Eli Lilly Japan KK, Taisho Pharma Co., Ltd., Terumo Corp., Tejin Pharma Ltd., Nippon Chemiphar Co. Ltd., Abbott japan Co. Ltd., and Johnson & Johnson K.K. Medical Co. and received honoraria from Nippon Boehringer Ingelheim Co. Ltd., Kissei Pharma Co. Ltd., Mitsubishi Tanabe Pharma Corp., Daiichi Sankyo Co. Ltd., Sanofi K.K., Takeda Pharma Co. Ltd., Astellas Pharma Inc., MSD K.K., Kyowa Kirin Co. Ltd., Sumitomo Dainippon Pharma Co. Ltd., Kowa Pharma Co. Ltd., Novo Nordisk Pharma Ltd., Ono Pharma Co. Ltd., Sanwa Kagaku Kenkyusho Co. Ltd., Eli Lilly Japan K.K., Taisho Pharma Co. Ltd., Bayer Yakuhin, Ltd., AstraZeneca K.K., Mochida Pharma Co. Ltd., Abbott Japan Co. Ltd., Teijin Pharma Ltd., Arkray Inc., Medtronic Japan Co. Ltd., and Nipro Corp., outside the submitted work. The other authors declare that they have no competing interests.
Author Contributions
MH led the drafting of the manuscript. YH and MF reviewed the manuscript and study design and contributed to the final draft. The other authors recruited the participants and contributed to the final draft.
Availability of Data and Materials
Data sharing is not applicable to this article, as no datasets were generated or analyzed during the current study.
Ethics Approval and Consent to Participate
The RISING-STAR study is registered with the Japan Registry of Clinical Trials (jRCTs051190114) and was approved by the ethics committees of the Kyoto Prefectural University of Medicine (CRB5180001). The RISING-STAR study is to be conducted in accordance with the Declaration of Helsinki. Written informed consent will be obtained from all the participants.
Patient and Public Involvement Statement
Patients were not involved in the design of the study, selection of research questions, or outcome measurements. Participants will not be involved in the interpretation or writing of the results. They will be given a simple summary of the study outcomes, written in Japanese, once the study has been completed.
Administrative Information
Note: the numbers in curly brackets in this protocol refer to SPIRIT checklist item numbers. The order of the items has been modified to group similar items (see ![]() ).
).
Protocol version: 1.0
Name and contact information for the trial sponsor: AstraZeneca K.K. (3-1, Ofukacho, Kita-ku, Osaka, Japan)
Ono Pharmaceutical Co. Ltd. (1-8-2, Kyutaromachi, Chuo-ku, Osaka, Japan)
Role of sponsor: SPIRIT guidance: Role of study sponsor and
The funders are not involved in study design; collection, management, analysis, and interpretation of data; writing of the report; and the decision to submit the report for publication, including whether they will have ultimate authority over any of these activities.
Trial Status
The RISING-STAR study started on March 2, 2020, with protocol version 1.1. Recruitment was completed on August 31, 2020.