
Abstract
Background and objective:
Over 2000 genotypes in the cystic fibrosis (CF) gene have been described. These genotypic differences result in variable clinical manifestations of CF, with severity of disease dependent on CF transmembrane conductance (CFTR) protein function. CFTR is widely distributed in nucleated cells, including cardiac myocytes, but the effect of genotype on cardiac function is not known.
Methods:
This retrospective review of echocardiographic data is from a single adult CF centre between 2000 and 2015. Patients were cohorted based on the functional classification of genotype. ‘Severe’ patients had both CF genes from functional classification groups 1-3; ‘mild’ patients had one or no gene from these groups, or in the event of the second gene being unknown were pancreatic sufficient.
Results:
Genotype and echocardiography were recorded during the inclusion period in 100 patients, 79 of whom were classified as having severe genotypes. Although the severe group were younger they had a lower fractional shortening (33.66
Conclusions:
This study is the first to show that in CF, severity of genotype (functional classification) is associated with cardiac impairment. Patients with severe CF genotype and cardiac dysfunction should be identified to evaluate cardiac response to gene-modifying treatments prior to consideration for lung transplantation.
Introduction
Cystic fibrosis (CF) is an autosomal, recessive disorder resulting from mutation of the cystic fibrosis trans-membrane conductance regulator (CFTR) gene, which causes a dysfunctional CFTR protein and apical cell-surface chloride ion channel. The consequences of this are multi-systemic and eventuate in well-characterised disease of the lung, exocrine pancreas, liver and sweat ducts. 1 Exercise capacity is impaired in CF, which is not solely explained by the reduced lung function and remains to be fully elucidated.2,3 Optimal exercise proficiency will be impaired by changes in lung function, cardiac function, intravascular volume or cell energetics. 4 Exercise tolerance can be significantly improved by treatments that potentiate the function of CFTR. 5
The identification of CFTR on human myocytes 6 has prompted investigation of a possible CF-related cardiomyopathy. Using the echocardiographic technique of longitudinal strain measurement, subclinical impairment in left ventricular function has been reported in a CF population.7,8 This finding is supported by earlier studies, which demonstrated exercise limitation related to a failure to augment cardiac output in patients with CF.9-11 It is unclear, however, whether abnormal cardiac function relates directly to pulmonary resistance, systemic sepsis, autonomic dysfunction or whether there is a direct effect of CFTR dysfunction on the cardiac myocyte. 12
There are over 2000 identified mutations of the CFTR gene, of which 276 are known to cause CF. 13 Mutations are classified based on the mechanism by which they disrupt CFTR function. Class 1, 2 and 3 mutations result from defective synthesis of full-length CFTR protein, defective CFTR protein processing and trafficking, and defective CFTR channel-gating, respectively. Classes 4 and 5 are a consequence of defective CFTR channel conductance or reduced synthesis/stability of CFTR protein, respectively. Whereas class 1-3 mutations typically show almost complete loss of CFTR function and severe CF disease, classes 4 and 5 mutations retain some CFTR activity and tend to result in milder phenotypes with longer survival.14,15 There is therefore a well-established precedence for categorising CF severity and prognosis on the basis of ‘low-risk’ or ‘high-risk’ genotype. 14 In this retrospective study in patients with CF, we hypothesised that patients with a high-risk genotype had greater cardiac dysfunction than those with a low-risk genotype based on echocardiographic findings.
Methods
Study design and patient population
This is a retrospective review of cardiac indices measured by transthoracic echocardiogram (TTE). Ethics approval was obtained from the institutional ethics committee at The Alfred Hospital (IEC46/18), and specific patient consent was not required due to the low risk nature of the study and collated de-identified data. Patients attended the Cystic Fibrosis Service, Alfred Health, Melbourne, Australia, and had a diagnosis of CF based on genotype and a positive sweat chloride. Patients with TTE performed as part of clinical care between 2000 and 2015 were included. Selection criteria were age ⩾ 18 years, no known history of cardiac disease or recent exacerbation at the time of echocardiography. The clinical record was reviewed from the time of TTE, and subjects were excluded if there was any clinical change or if physiologic support was required (oxygen supplementation, ventilation, fluid resuscitation or inotropic/vasopressor agents).
Collected data included demographic details, genotype, FEV1, laboratory data and echocardiographic indices of cardiac function. Laboratory results performed within 60 days and closest temporally to the date of TTE were recorded. The most recent FEV1 to the date of echocardiography was also recorded.
Subjects were classified into 2 genotype groups on the basis of functional class of identified CFTR allele mutations. Patients were considered to have a severe genotype if they had 2 CFTR mutations from classes 1-3. Patients with only one or no class 1-3 mutations were considered to have a mild phenotype. When a second allele mutation was not identified, patients were only included in the analysis if they were documented to be pancreatic sufficient (suggestive of a milder clinical phenotype) at transition to adult CF care.
Echocardiography measurements
Echocardiographic studies were performed (iE33; Philips Healthcare, North Ryde, New South Wales, Australia) with subjects examined in a semi-supine, left-lateral position. The electrocardiogram was recorded continuously. Global left ventricular function was assessed from an apical four-chamber view by measuring left ventricular ejection fraction using the modified monoplane Simpson’s rule.
Left ventricular end-diastolic diameter (LVDD), left ventricular end-systolic diameter (LVSD), right ventricular end-diastolic diameter, and right ventricular anterior wall thickness were measured from the M mode from a left parasternal view. Left ventricular fractional shortening (FS) was calculated from the formula (LVDD – LVSD/LVDD) X 100. Left atrial area (LA area) and right atrial area were measured from the apical four-chamber view. LA volume (LA volume) was calculated using the biplane area-length method and normalised to body surface area. The mitral Doppler signal was recorded in the apical four-chamber view, with the Doppler sample volume placed at the tip of the mitral valve.
The peak velocities of early (E) and late (A) filling waves and early/late filling ratios of peak velocities (E/A) were measured on the basis of trans-mitral flow velocities. Pulmonary systolic artery pressure was estimated from continuous-wave Doppler of tricuspid regurgitation using the Bernoulli equation. Tricuspid regurgitation velocity was recorded from the apical view and the parasternal short-axis view. Right atrial pressure (RAP) was estimated by assessing the diameter of the inferior vena cava and the percentage decrease in its diameter during inspiration. Tricuspid Annular Plane Systolic Excursion (TAPSE) was recorded on M-mode using the apical four-chamber view with the cursor is placed at the junction of the tricuspid valve plan with the free wall of the right ventricle.
Statistical analysis
Statistical analysis was performed using GraphPad InStat version 3.10 for Windows (GraphPad Software, San Diego, California, USA). Mean and standard deviation were calculated for comparison of demographic and laboratory data. Comparisons of echocardiographic indices of cardiac function were made between patients with severe and mild CFTR genotype configurations using the student T-test. Pearson’s correlation coefficients were calculated to assess associations between indices of left ventricular function and recorded variables.
Results
Between the years 2000 and 2015, a TTE was performed in 100 genotyped patients (Table 1). The study was performed to exclude pulmonary hypertension (n = 17), valve dysfunction (n = 20), endocarditis (n = 6), and causes for arrhythmias (n = 7), neurological events (n = 10) or as part of routine lung transplant workup (n = 40). Patients were 31.5 ± 11.2 years old and had moderately severe lung disease (FEV1 47.3 ± 23.4% predicted) but without pulmonary hypertension (RVSP 36.1 ± 12.4 mmHg). Mean left ventricular ejection fraction (LVEF) was low to normal (61.7 ± 7.4%).
Patient characteristics and echocardiographic indices of cardiac function for total cohort and by CFTR genotype severity.

BSA: body surface area; FEV1: forced expiratory volume in 1 second; LVDD: left ventricular diastolic diameter; LVSD: left ventricular systolic diameter; FS: left ventricle fractional shortening; LVEF: left ventricular ejection fraction; LA: left atria; E/A: mitral ratio of peak early to late diastolic filling velocity; RVSP: right ventricular filling pressure; TAPSE: tricuspid annular plane systolic excursion; RAP: right atrial pressure.
One hundred patients had at least one CF-causing CFTR mutation identified and were studied in the primary analysis by genotype severity (Table 1). Seventy-nine patients had mutations of both alleles from the CFTR classification groups 1-3 (severe genotype), and 21 had a single allelic mutation (milder genotype). Of the 21 patients with mild genotype, 13 did not have a second CFTR mutation identified. Patients with a severe genotype were younger than those with a mild genotype (31.1 ± 10.0, 44.0 ± 16.8 years; P < .0001).
No significant difference was observed between patients with severe and mild genotype in right ventricular systolic pressure (RVSP) (32.9 ± 9.4 mmHg versus 35.8 ± 9.4 mmHg; P = .33) or FEV1 (45.2 ± 22.6% versus 51.8 ± 25.1% predicted; P = .26). There was a wide range of FEV1 in both groups; however, other factors such as effective airway clearance, medication compliance and influence of the unknown gene can affect FEV1 performance. The severe genotype group had a significantly lower FS (33.3 ± 6.6 versus 36.9 ± 6.3 %; P < .05) and a lower but non-significant LVEF (60.5 ± 6.3 versus 63.5 ± 4.9%; P = .15). They also had a lower LA area (14.9 ± 3.6 versus 18.0 ± 4.2 cm2; P < .01) and LA volume (39.9 ± 18.7 versus 51.0 ± 18.7 mL; P < .05).
Univariate analysis using Pearson’s correlation identified a linear association between FS and TAPSE (r = 0.2726, P < .05); however, there was no association between FS and LA area, LA volume or FEV1 (Table 2).
Univariate analysis of fractional shortening.
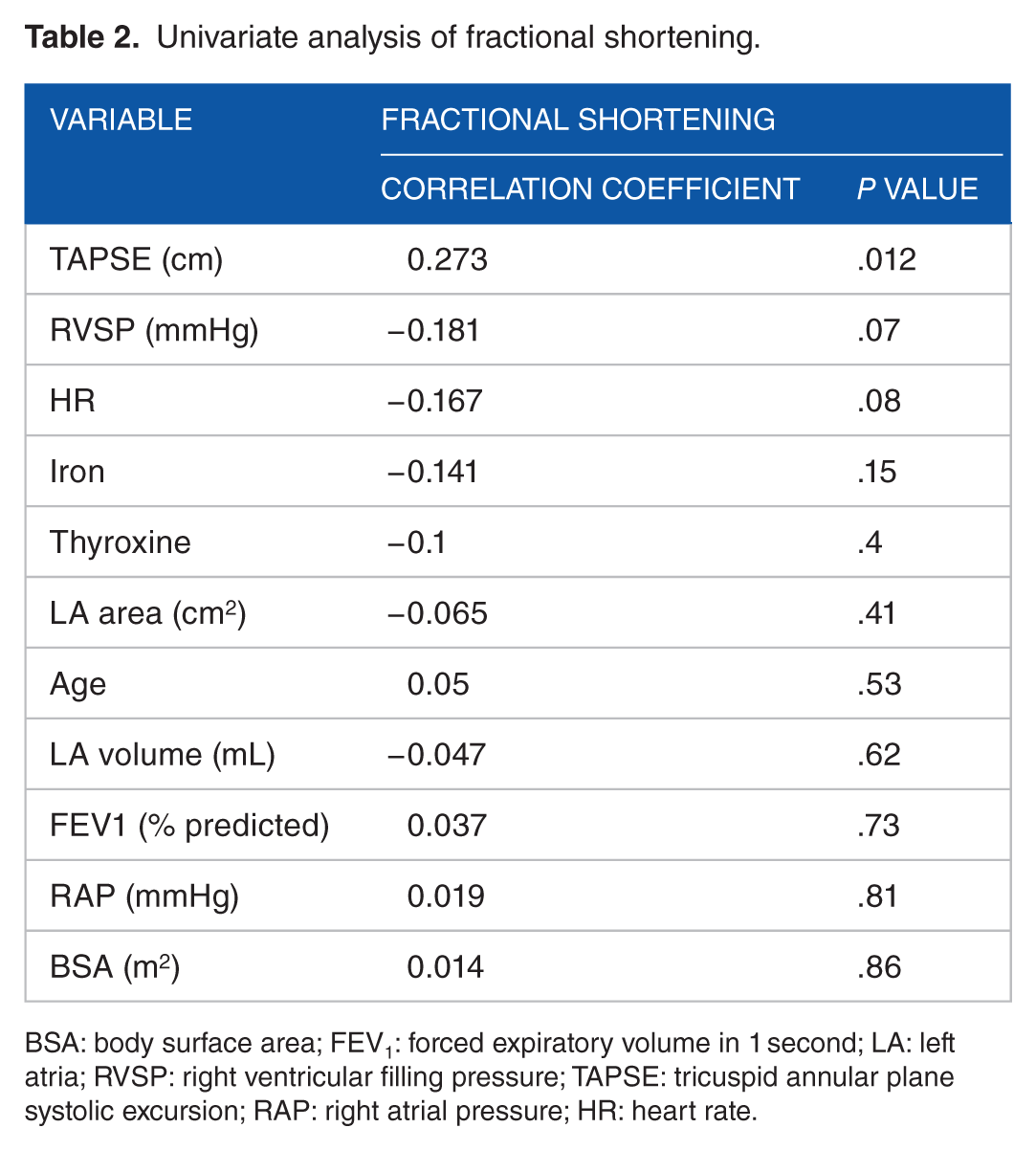
BSA: body surface area; FEV1: forced expiratory volume in 1 second; LA: left atria; RVSP: right ventricular filling pressure; TAPSE: tricuspid annular plane systolic excursion; RAP: right atrial pressure; HR: heart rate.
Discussion
In this study, we found that CF patients with a severe genotype had a lower left ventricular fractional shortening and TAPSE than their counterparts with a mild genotype. This finding was independent of lung function severity based on ppFEV1 or nutritional changes; however, the specific mechanism by which classification 1-3 CFTR mutations might effect change in cardiac function was not part of this study.
Previous studies show left atrial dimensions reflect volume status, 20 and here, the lower LA area and LA volume in the severe genotype group favours a hypovolaemic state, previously reported in CF, 21 causing a decrease in preload and reducing FS. The trend towards a negative association between heart rate and FS also supports decreased preload as a potential mechanism. Systolic and diastolic blood pressure (BP) between both groups did not differ implying afterload changes were either not influenced by genotype differences or compensatory mechanisms were able to moderate their impact at rest. Potential confounders for altered FS might be myocardial fibrosis, although no regional wall changes were seen on Echo, while autonomic dysfunction in CF is not common. 12 TAPSE was also lower (although within normal range) in the severe group identifying early RV changes, while LVEF did not differ between groups. Huang et al 22 have recently showed that single plane assessment was more sensitive than volumetric data in an intensive care unit (ICU) setting where volume changes may affect measurement.
Normal CFTR function is necessary for optimal cardiac function. Research has shown that in failing hearts, the CFTR adenosine triphosphate (ATP) binding cassettes are almost completely absent. 23 Murine studies describe reduced cardiac contraction by CFTR inhibition and blunting of the effect with stimulation of calcium-activated chloride channels. 24 Impaired CFTR activity on cardiac myocytes in patients with CF might directly result in subclinical cardiac dysfunction either through a direct myocardial impairment or a remodelling effect after injury. 25 Poor expression of mature CFTR protein has been reported in heart failure patients without CF in comparison to control subjects suggesting CFTR deficiency has a role not only in depolarisation but also contractility. 24 One potential explanation for the role of CFTR might be that in response to strain (failing respiratory function, or cardiac ischaemia) normally functioning chloride channels assist in repair of injured myocardium. Genetically or functionally (antibiotics, smoking) hindered CFTR may result in myocardial dysfunction.
The use of strain rate echocardiography in CF patients has unmasked subclinical LV dysfunction not present on resting techniques.7,8 These findings have also been supported by work using equilibrium radionuclide cineangiography. 9 Exercise also exposes CF cardiac dysfunction; using an acetylene rebreathe technique during cardio pulmonary exercise testing (CPET), Van Iterson et al 11 showed impaired cardiac function contributed to CF exercise limitation. Adrenergic stimulation itself may also induce dysfunction in primed CFTR-defective myocardium, with evidence that albuterol (selective B2-agonist) impaired cardiac output (6.7 +/– 0.5 vs 9.1 +/– 0.3 mL/min/m2) compared to healthy individuals. 26
The difference in FS and TAPSE between high-risk and low-risk CF genotypes indicates a change in myocardial function as identified in the literature; 27 however, the variability of this effect based on genotype has not been identified. We know that severe genotypes are associated with less CFTR activity than milder genotypes and have reduced survival. 14 A recent retrospective analysis of the United States’ Cystic Fibrosis Foundation (CFF) patient registry (25,753 patients) found sweat chloride concentration at CF diagnosis predicted long-term survival, due mainly to the differences in CFTR genotype. Patients with residual CFTR function (classification 4-5) had better survival compared with CFTR genotypes with severely reduced CFTR function (classification 1-3). 14
There are some limitations to the current study. This is a retrospective study, and cardiorespiratory stability is difficult to assess accurately as this relied on review of physiological variables in the patient’s medical record, while identifying contemporaneous FEV1 with echocardiography is also limited by this design. However, these values are assessed regularly in the routine care of adult CF patients limiting the time lag. In 13 subjects, the second CFTR mutation was unidentified, it is unlikely but possible that the second mutation was a class 1-3 mutation in these 13 patients which may influence the findings. We have minimised this limitation in these subjects by only including those with pancreatic sufficiency at transition to adult CF care in the mild genotype group.
There is a growing appreciation that CF is associated with subclinical ventricular dysfunction. In this study, we found a greater compromise in cardiac function associated with severe CF genotypes versus mild genotypes, while severity of lung disease was unrelated. Recently, CFTR potentiator and corrector treatment has improved patient lung function and quality of life; however, the impact on cardiac disease in CF is unknown. Further clinical trials are needed to delineate the impact of cardiac CFTR dysfunction on CF patients and if CFTR modulators might improve cardiac function in CF patients with severe genotypes.
Footnotes
Funding:
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ Note
The authors M.P., D.T.K., D.M.K., T.K., and J.W. have no financial relationships or conflicts of interest to declare with respect to this manuscript.