
Abstract
Introduction:
Burkitt lymphoma (BL) is a highly aggressive B-cell non-Hodgkin lymphoma that typically presents with abdominal masses; ampullary involvement with obstructive jaundice and gastrointestinal bleeding is exceptionally rare in children. Early biliary decompression can be crucial to permit timely chemotherapy.
Case presentation:
A 14-year-old boy presented with 1 week of worsening abdominal pain, non-bilious vomiting, constipation, jaundice, and melena. Examination showed pallor, icterus, firm hepatomegaly, and a palpable epigastric mass. Laboratory testing revealed cholestatic liver function abnormalities and elevated pancreatic enzymes. Ultrasound and contrast computed tomography demonstrated a large retroperitoneal mass compressing the biliary tree. Upper gastrointestinal endoscopy identified a friable, ulcerated ampullary mass with active bleeding. Biopsy confirmed BL by morphology (starry-sky) and immunohistochemistry/fluorescence in situ hybridization (CD20+, CD10+, c-MYC+, Ki-67 ~95%). Main diagnosis made was pediatric BL presenting with ampullary involvement causing obstructive jaundice and upper gastrointestinal bleeding. Given persistent cholestasis and bleeding risk, the patient underwent Roux-en-Y choledochojejunostomy for biliary decompression, followed by initiation of rituximab–cyclophosphamide–vincristine–doxorubicin–high-dose methotrexate/rituximab–ifosfamide–etoposide–high-dose cytarabine (R-CODOX-M/R-IVAC) with central nervous system prophylaxis. Post-operative bilirubin improved, and early chemotherapy cycles were tolerated. During the 2-week hospitalization, the patient had symptomatic improvement; however, objective radiologic response could not be documented because care was transferred and post-transfer positron emission tomography–computed tomography was unavailable.
Conclusion:
Ampullary BL should be considered in pediatric patients with obstructive jaundice and upper gastrointestinal bleeding. Surgical biliary decompression can stabilize cholestasis and facilitate timely multi-agent chemotherapy.
Keywords
Introduction
Burkitt lymphoma (BL) is a highly aggressive high-grade B-cell Non-Hodgkin Lymphoma (NHL) and one of the most common pediatric cancers worldwide. According to the Global Burden of Disease estimates, childhood non-Hodgkin lymphoma accounted for ~20 800 new cases and 12 700 deaths globally in 2021, with BL representing a major subtype. 1 The endemic variant, strongly linked to Epstein–Barr virus and malaria, predominates in equatorial Africa with jaw and facial bone involvement, whereas the sporadic form is typical in Western countries, 2 In Pakistan, BL is likewise an important pediatric malignancy. Data from a major tertiary-care center identified BL as the most common subtype of childhood NHL (35%-40%), frequently presenting with abdominal masses and advanced-stage disease. 3 Despite this, outcomes remain poor, as cancers overall contribute to ~12% of total deaths nationally, with lymphoma representing a substantial share. 4
The abdomen is considered the hallmark site of sporadic BL, but even within this distribution, the periampullary region remains vanishingly rare. We report the case of a 14-year-old boy with BL presenting as an ulcerated ampullary mass mimicking adenocarcinoma, underscoring the diagnostic pitfalls and anatomical unpredictability of sporadic BL and contextualizing its systemic manifestations. This case has been reported following the CARE criteria (Supplemental Material). 5
Case Presentation
A 14-year-old male, born of a non-consanguineous marriage, presented with a history of progressively worsening abdominal pain for 8 to 10 days, associated with repeated episodes of non-bilious vomiting and constipation. Parents noticed yellowish discoloration of the eyes and skin over the same duration. The pain was initially mild but became severe during the week prior to admission. There was no associated fever, loose stools, hematemesis, or history of oral ulcers, weight loss, or night sweats. Past medical and surgical history was unremarkable, and there was no history of tuberculosis, HIV, or chronic illness. Family history was non-contributory. The child lives in a low-income household with a single breadwinner, consuming untreated tap water. During hospitalization, the patient developed melena with a rapid fall in hemoglobin from 12.8 to 5.5 g/dL within 24 hours. He received 2 packed cell transfusions with partial improvement in hemoglobin, which remained unstable throughout the hospital stay. The patient’s clinical course is summarized in a timeline (Figure 1).

Clinical course timeline.
Examination
On admission, he was hemodynamically stable with a blood pressure of 110/84 mmHg, pulse 92/min, respiratory rate 18/min, and Body Mass Index within normal range. General physical examination revealed pallor and icterus. No peripheral lymphadenopathy, clubbing, or edema was noted. Abdominal examination revealed firm, tender hepatomegaly with right lobe span of approximately 14 cm and a palpable epigastric mass. Splenomegaly was not appreciable. Cardiovascular, respiratory, and neurological examinations were unremarkable. Genitourinary examination was significant for dysuria and frequency.
Laboratory Investigations
The patient’s blood counts showed a progressive fall in hemoglobin, requiring transfusions. Liver function tests showed direct hyperbilirubinemia. Lactate Dehydrogenase was elevated, consistent with high tumor burden. Laboratory Investigations conducted are summarized in Table 1.
Laboratory Investigations During Hospital Course.

Abbreviations: anti-HCV, hepatitis C virus antibody; HBsAg, hepatitis B surface antigen; HIV, human immunodeficiency virus.
Initial Differential Diagnosis
Based on the presenting features of jaundice, abdominal mass, and raised pancreatic enzymes, initial differentials considered included liver abscess, hydatid cyst, acute hepatitis, urinary tract infection, and pancreatitis. Extrahepatic biliary obstruction secondary to neoplasm was suspected after imaging.
Imaging
Abdominal ultrasound (US) revealed a sludge-filled gallbladder with a large heterogenous retroperitoneal mass (7.6 × 7.0 cm) and multiple enlarged para-aortic and peripancreatic lymph nodes (up to 1.6 × 1.5 cm). US findings could not be displayed; see Limitations. Computed Tomography (CT) abdomen (Figure 2) with contrast demonstrated a large lobulated, homogeneously enhancing retroperitoneal mass crossing the midline, displacing the aorta, encasing renal arteries, and invading hepatic segments I, VI, and VII. The mass compressed the cystic duct with resultant intrahepatic biliary dilatation, extended to the right renal hilum causing mild hydroureteronephrosis, and abutted the prevertebral space without bony erosion. Multiple mesenteric and para-aortic lymph nodes were present, and free pelvic fluid was noted. CT chest was normal.
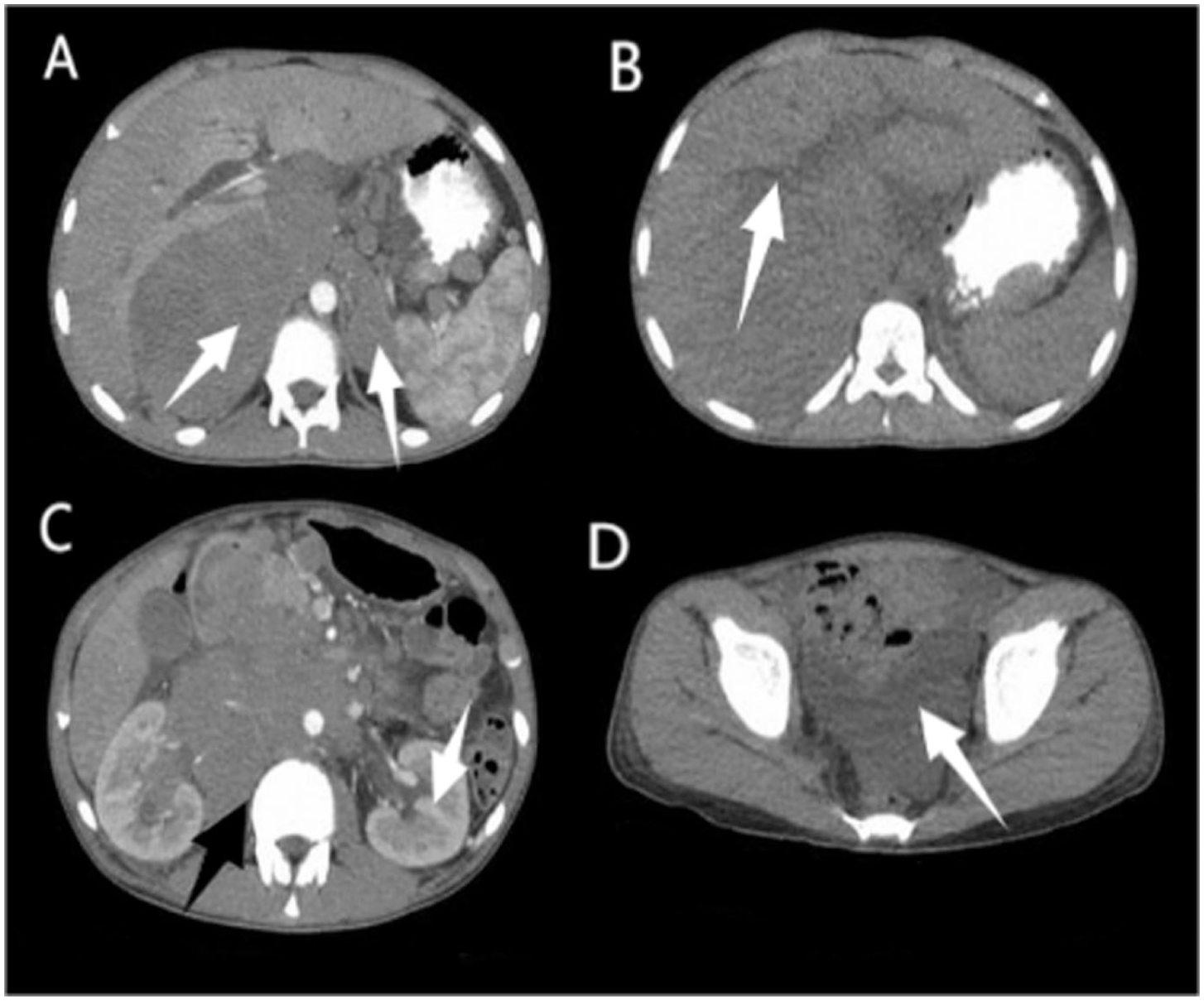
(A) Contrast-enhanced CT axial showing a large lobulated retroperitoneal soft-tissue mass, measuring approximately 8.9 × 15.2 × 16.7 cm in anteroposterior, transverse and coronal planes respectively, encasing the aorta and displacing adjacent bowel loops, (B) non-contrast CT axial section demonstrating the mass closely abutting the porta hepatis with focal loss of fat planes, causing mild proximal intrahepatic biliary dilatation, (C) contrast CT axial section at the level of the left renal hilum shows the mass displacing the renal hilum and causing mild left-sided hydroureteronephrosis (white arrow). The lesion is also seen closely abutting the prevertebral space and paravertebral musculature (black arrow), and (D) non-contrast CT axial section of the pelvis demonstrating small volume free intraperitoneal fluid.
Endoscopy
Upper Gastrointestinal (GI) endoscopy (Figure 3) revealed an ulcerative, friable ampullary mass compressing the duodenal lumen with clots and active bleeding. The esophagus and stomach were normal except for blood-streaked gastric contents.

Endoscopic images of the ampulla of Vater: (A-C) wide views demonstrating an ulcerated ampullary mass and (D) close-up view of the lesion with contact bleeding.
Histopathology and Molecular Studies
Biopsy from the ampullary mass showed sheets of high-grade lymphoid cells with a “starry-sky” pattern. Immunohistochemical (IHC) staining was performed on formalin-fixed paraffin-embedded tissue sections using commercially available antibodies. Fluorescence in situ hybridization (FISH) was performed to confirm the diagnosis using a dual-color break-apart probe. IHC, FISH, and molecular profile findings have been displayed in Table 2. These findings, in conjunction with morphology and immunophenotype, supported the diagnosis of Burkitt lymphoma.
Integrated Immunohistochemical and Molecular Profile of Ampullary Mass Biopsy Consistent with Burkitt’s Lymphoma.

Treatment and Clinical Course
The patient was initially managed with intravenous antibiotic meropenem 1 g IV q8h, Acetaminophen 500 mg PO q6h PRN (daily total dose kept below 3 g in view of cholestasis), hydration, IV N-acetylcysteine protocol 150 mg/kg over 1 hour, then 50 mg/kg over 4 hours, then 100 mg/kg over 16 hours, ursodeoxycholic acid 13 mg/kg/day for cholestatic symptoms N/S IV and packed red cell transfusion using a restrictive strategy (Hb < 7 g/dL); all due to the working diagnoses mentioned previously. Despite supportive care, symptoms persisted and anemia worsened. Following histopathological confirmation, the patient was referred to oncology.
In view of progressive obstructive jaundice, persistent hyperbilirubinemia (total bilirubin 7.7 → 6.4 mg/dL with direct predominance), and concern for cholangitis, a surgical biliary decompression was undertaken. At laparotomy, a large ampullary mass was identified causing external compression of the distal common bile duct and duodenum. The liver and gallbladder were grossly distended, but no focal hepatic lesions were seen. Intraoperative frozen section confirmed high-grade lymphoid proliferation consistent with lymphoma. A Roux-en-Y choledochojejunostomy was performed to establish biliary drainage. Intraoperatively, careful dissection was required due to adherence of the mass to the pancreatic head and mesenteric vessels; no attempt was made at debulking, given the high chemosensitivity of Burkitt lymphoma and risk of delaying systemic therapy. The postoperative course was uneventful, with decline in serum bilirubin over 5 days, improved appetite, and resumption of enteral nutrition. The patient was subsequently referred for definitive oncologic management. Rituximab, cyclophosphamide, vincristine, doxorubicin, high-dose methotrexate/ifosfamide, etoposide, high-dose cytarabine (R-CODOX-M/R-IVAC) with Central Nervous System (CNS) prophylaxis chemotherapy regimen was followed and is described in Table 3.
Oncology Regimen for Burkitt Lymphoma (R-CODOX-M/R-IVAC).

Follow-Up Clinical Course
The patient was discharged after a 2-week hospitalization and referred for definitive oncological management. Subsequently, the patient transferred care to another center. Despite repeated attempts, records from the receiving institution, including restaging studies, were unavailable at the time of manuscript preparation. As for the last verified contact from our center, no objective post-transfer disease assessment had been documented. Accordingly, remission status within 2 months post-surgery cannot be confirmed; See Limitations.
Patient Perspective
The patient’s caregivers reported significant distress during the period of painless jaundice and diagnostic uncertainty. They expressed relief after receiving a definitive diagnosis and following biliary bypass, which alleviated pruritus and improved oral intake. They valued clear counseling about the urgency of initiating rituximab-containing, multi-agent chemotherapy and CNS prophylaxis. The decision to transfer care to a better equipped institution reduced logistical burdens but created challenges obtaining records and imaging for the doctors at the original center, which the family understood may limit what can be reported here. Overall, they emphasized timely communication and coordination as the most helpful aspects of care.
Discussion
Clinical Misdirection
Patients with small intestine lymphoma may experience a wide range of symptoms, including nonspecific abdominal discomfort, nausea, vomiting, and weight loss. A rare symptom of small intestine lymphoma is obstructive jaundice. On endoscopy, small intestine lymphoma can appear as a mass, polyp, or ulcer that is difficult to differentiate from carcinomas. The presence of melena in ampullary Burkitt lymphoma is highly unusual and prompted a biopsy to rule out carcinoma. 6 The systemic manifestations required a vigorous and brisk treatment plan on boarding oncology and general surgery.
The massive retroperitoneal and periampullary mass discovered on CT compressed the biliary tree, resulting in direct hyperbilirubinemia, jaundice, and cholestatic liver function abnormalities. The displacement of surrounding structures by the mass led to mild hydroureteronephrosis, which in turn caused dysuria and urinary frequency along with abdominal pain. Endoscopic examination revealing an ulcerative friable ampullary mass with active bleeding explains the cause of melena and anemia. Together, these radiographic and endoscopic findings explain the patient’s lab abnormalities and symptom progression, emphasizing the mass’s aggressive local behavior and tendency to mimic more common periampullary cancers.
Diagnostic Challenges
Differentiating BL from other high-grade B-cell lymphomas, particularly diffuse large B-cell lymphoma (DLBCL), poses a well-recognized diagnostic challenge due to overlapping histologic and immunophenotypic features. In this case, the combination of morphologic findings, characteristic immunoprofile, and cytogenetic confirmation provided conclusive evidence of BL. The biopsy demonstrated a monomorphic population of medium-sized lymphoid cells with multiple basophilic nucleoli and interspersed tangible-body macrophages, producing a classic “starry-sky” appearance. Immunohistochemistry revealed CD20 and CD10 positivity, confirming B-cell and germinal center origin. BCL6 positivity further supported germinal center derivation with violent biological behavior, while absence of BCL2 expression argued against DLBCL or double-hit lymphoma. The extremely high Ki-67 proliferation index (~95%) reflected the near-total growth fraction characteristic of BL, distinguishing it from DLBCL, where the index is typically lower and more heterogeneous. It was necessary to confirm the results with evidence of C-MYC gene rearrangement through FISH. Without cytogenetic validation, the appearances were most likely to be a high-grade B-cell lymphoma with Burkitt lymphoma in the differential. FISH confirmed a MYC rearrangement at 8q24 with no BCL2 or BCL6 co-rearrangements, excluding double-hit or triple-hit high-grade B-cell lymphomas. Additionally, the unmutated immunoglobulin heavy-chain variable region (VH) gene indicated a germinal center B-cell origin with minimal somatic hypermutation, further supporting the diagnosis of BL. As described by previous studies, endoscopic biopsy combined with immunohistochemistry (CD20, CD10 positive, increased MYC, increased Ki-67, lack of BCL2 rearrangements), cytogenetics, and flow cytometry remains definitive for accurate diagnosis. 7 Collectively, these findings established Burkitt’s lymphoma as the final diagnosis.
Management Challenges
BL is a rapidly growing, aggressive malignancy with distinct clinical problems. Treatment was based on the high-dose chemotherapy regimen R-CODOX-M/R-IVAC with CNS prophylaxis. According to a phase 3 trial published by the Lancet, this regimen is associated with hazards such as myelosuppression, infection, mucositis, renal and cardiac toxicity, and neurotoxicity. Supportive care—hydration, Tumor Lysis Syndrome (TLS) prophylaxis, transfusions, antiemetics, antimicrobials, and monitoring of organ function—is crucial for safe and effective treatment.6,8 Given the patient’s age, conventional therapy and standard protocols were followed without major dose modification. Obstructive jaundice is an uncommon but serious BL complication, typically resulting from biliary infiltration or compression by tumor masses. Hyperbilirubinemia interferes with drug metabolism, at times requiring dose adjustment or treatment delay, hence impairing prognosis. Endoscopic or percutaneous biliary drainage is preferred, with surgical bypass reserved when less invasive procedures fail or when cholangitis risk is high as was required in this patient. 9 The “field effect” of the tumor mass resulting in multi-system pathology was a major concern in this case. It warranted a holistic management plan factoring in the age and financial status of the patient. The general volatile situation of patients presenting to Pakistani Tertiary care government hospitals makes leaving against medical advice mid-management more likely.
A schematic displaying the treatment outcomes and modalities is presented in Figure 4. The case reports of pediatric and young-adult patients provide evidence that a restricted surgical procedure can be followed with intensive chemotherapy, resulting in favorable outcomes—with surgery serving as an adjuvant or facilitating measure rather than curative therapy. 10 When BL is suspected pre-operatively, some centers advocate initial cytoreductive chemotherapy or a short cyclophosphamide–vincristine–prednisone (COP) pre-phase before full-intensity treatment or employ a brief corticosteroid window for urgent tumor cytoreduction. Such medical debulking can induce rapid regression and occasionally obviate the need for extensive surgery when tissue diagnosis is already secured. 11 However, in this case the child’s severe obstructive jaundice and markedly elevated bilirubin posed a prohibitive risk for early chemotherapy because of potential drug-induced hepatotoxicity and exacerbation of cholestasis. Under these circumstances, timely biliary decompression is both guideline-supported and evidence-based, as reflected in reports describing surgical and endoscopic drainage for lymphoma-related biliary obstruction.10,12 Moreover, recent literature on emergency management of complicated intestinal lymphoma supports limited, goal-directed surgery as appropriate when necessary to stabilize the patient or enable safe chemotherapy. 13 Hence, early surgical intervention was the optimal management, functioning as a prerequisite step that allowed the prompt commencement of curative multi-agent immunochemotherapy—fully consistent with contemporary pediatric BL treatment principles.10-13
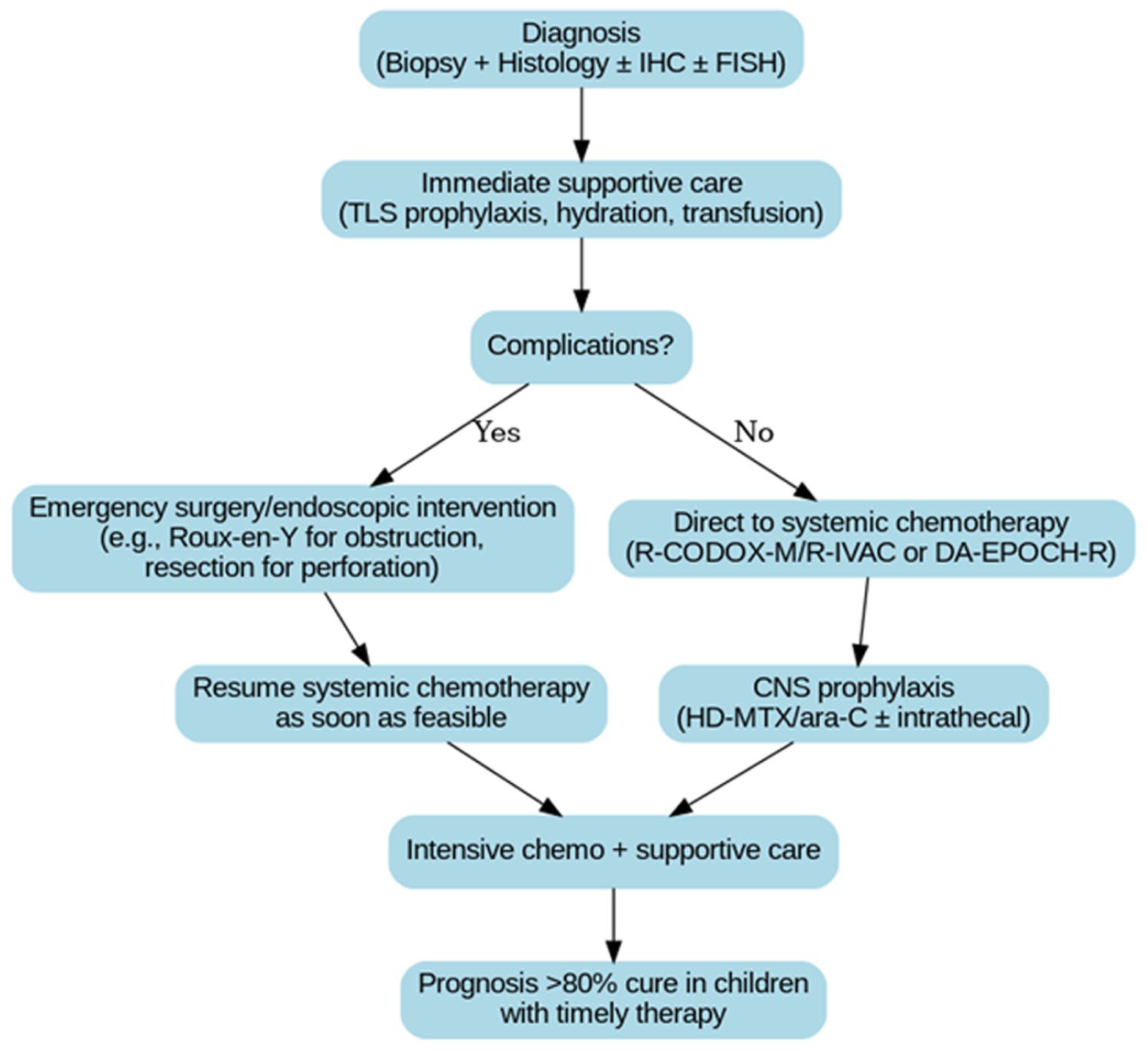
Management algorithm for gastrointestinal Burkitt’s lymphoma demonstrating the role of limited surgery for biliary decompression followed by curative systemic chemotherapy.
Pathophysiology
The hallmark of Burkitt lymphoma is a translocation of the c-MYC gene on chromosome 8. Around 80% of all Burkitt lymphomas exhibit a t(8:14) translocation. The translocation of the MYC oncogene to the promotor sequence of the IgG heavy chain gene leads to constitutive activation regardless of the specific translocation. Given that activation-induced cytidine deaminase (AID) is necessary for MYC translocation and that AID is strongly expressed in the germinal center, it is believed that BL originates from germinal center B cells. Since c-myc regulates the expression of numerous genes, its constitutive expression results in cell proliferation, uncontrolled growth and a lowered apoptotic threshold.14,15 Primary lymphomas of the GI tract are uncommon neoplasms, and they occur primarily in the stomach. In children, primary gastric lymphoma accounts for 1.48% of all stomach malignancies. Involvement of the duodenum is primarily uncommon due to the sparsity of lymphoid tissue in the region. It accounts for even less than 5% of all lymphomas of the small bowel. The lymphoma of the ampulla of Vater is even rarer with only a handful of cases in literature. This is the first case that presented as an ampullary mass with GI bleeding, biliary dilation and related symptoms, and hydroureteronephrosis in any major tertiary care setup in the country. One case from Pakistan which has been mentioned was protein-losing enteropathy-focused and did not compress the ampulla of Vater.6,16-19 Review of similar cases is summarized in Table 4.
Summary of Similar Published Pediatric Gastrointestinal Burkitt Lymphoma Cases.
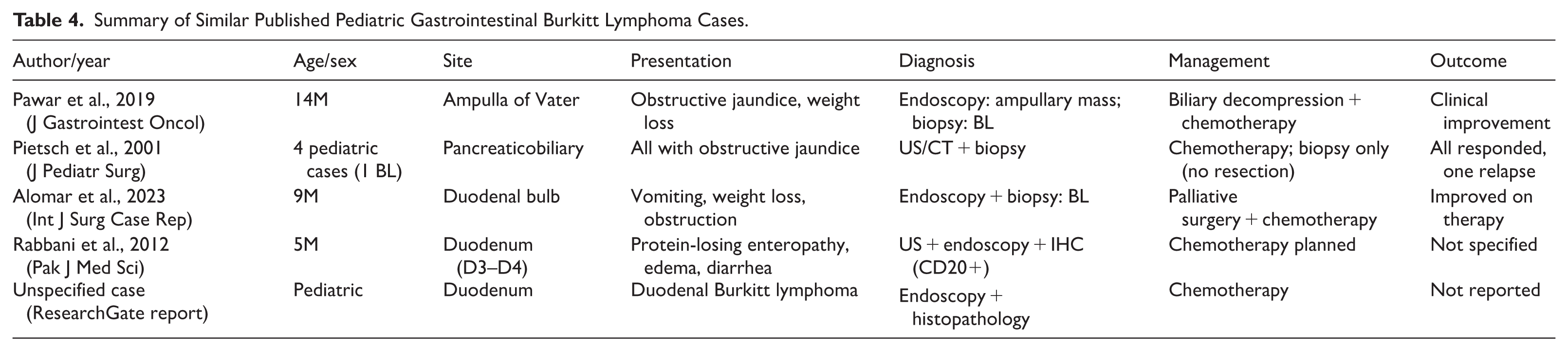
A review of previously reported cases of pancreaticobiliary Burkitt lymphoma identifies recurrent patterns. Almost all cases consisted of boys between the ages of 5 and 14 with obstructive jaundice caused by involvement of the periampullary region or the pancreatic head. The lesions were usually confined, with no multi-compartment retroperitoneal dissemination or gastrointestinal hemorrhage. Endoscopy showed non-ulcerative lesions, and pancreatic enzyme levels were normal or not reported. Burkitt’s lymphoma morphology and immunophenotype (CD20⁺, CD10⁺, BCL6⁺, Ki-67 > 95%, MYC) were used for diagnosis in all cases. Management typically favored biopsy with little surgical intervention followed by aggressive chemotherapy, which resulted in positive outcomes.
The presentation with melena during hospitalization accompanied by a rapid drop in hemoglobin, endoscopy showing an ulcerated friable ampullary lesion with active bleeding, markedly elevated pancreatic enzymes mimicking pancreatitis, a massive retroperitoneal lesion crossing the midline encasing major vessels and causing hydroureteronephrosis, molecular workup including VH gene mutation testing, a Roux-en-Y choledochojejunostomy performed due to the increased risk of cholangitis before initiating chemotherapy; are all features unique to this case.
Adult versus Pediatric Age Groups for GI Non-Hodgkin’s Lymphoma
Gastrointestinal non-Hodgkin lymphomas (GI NHLs) are predominantly diseases of middle-aged and older adults. Most large series place the median age of diagnosis in the late 50s to early 60s. For example, in a cohort of 415 adult patients, the median age was 57 years, and DLBCL accounted for the majority of cases. 20 Subtype-wise, Mucosa-Associated Lymphoid Tissue (MALT) lymphomas typically occur in later adulthood, clustering around the fifth to seventh decades, while DLBCL remains the dominant aggressive histology in this age group. T-cell lymphomas, such as enteropathy-associated types, are much rarer but likewise show a strong older-adult predilection. 21 In a separate Indian retrospective analysis of 152 GI lymphomas, the adult subgroup (n = 133, ⩾18 years) demonstrated: DLBCL 54%, Burkitt 19%, MALT 11%, and other subtypes (mantle cell, plasmablastic, T-cell, follicular) 16%. 22
In contrast, pediatric and adolescent GI NHL occur primarily in childhood and the teenage years, with a very different subtype distribution. In a population-based Surveillance, Epidemiology, and End Results study of 334 patients, the proportions were: Burkitt lymphoma 56.9%, DLBCL 27.8%, other rare subtypes 15.3%, and MALT 0%. 23 Other indolent histologies, such as MALT, are essentially absent in children. Smaller clinicopathological reviews confirm that GI NHL represents <5% of pediatric cancers overall, but within this group, the burden falls on school-aged children and adolescents, especially in the form of aggressive Burkitt lymphoma. 24 The comparative analysis of subtype distribution is provided visually in Figure 5. Diagnostic confusion with ampullary carcinoma occurs in both age groups because pediatric and adult ampullary lymphoma exhibit typical radiologic and clinical characteristics, such as obstructive jaundice, stomach pain, and occasionally weight loss which traverse a wide range of differential diagnoses.
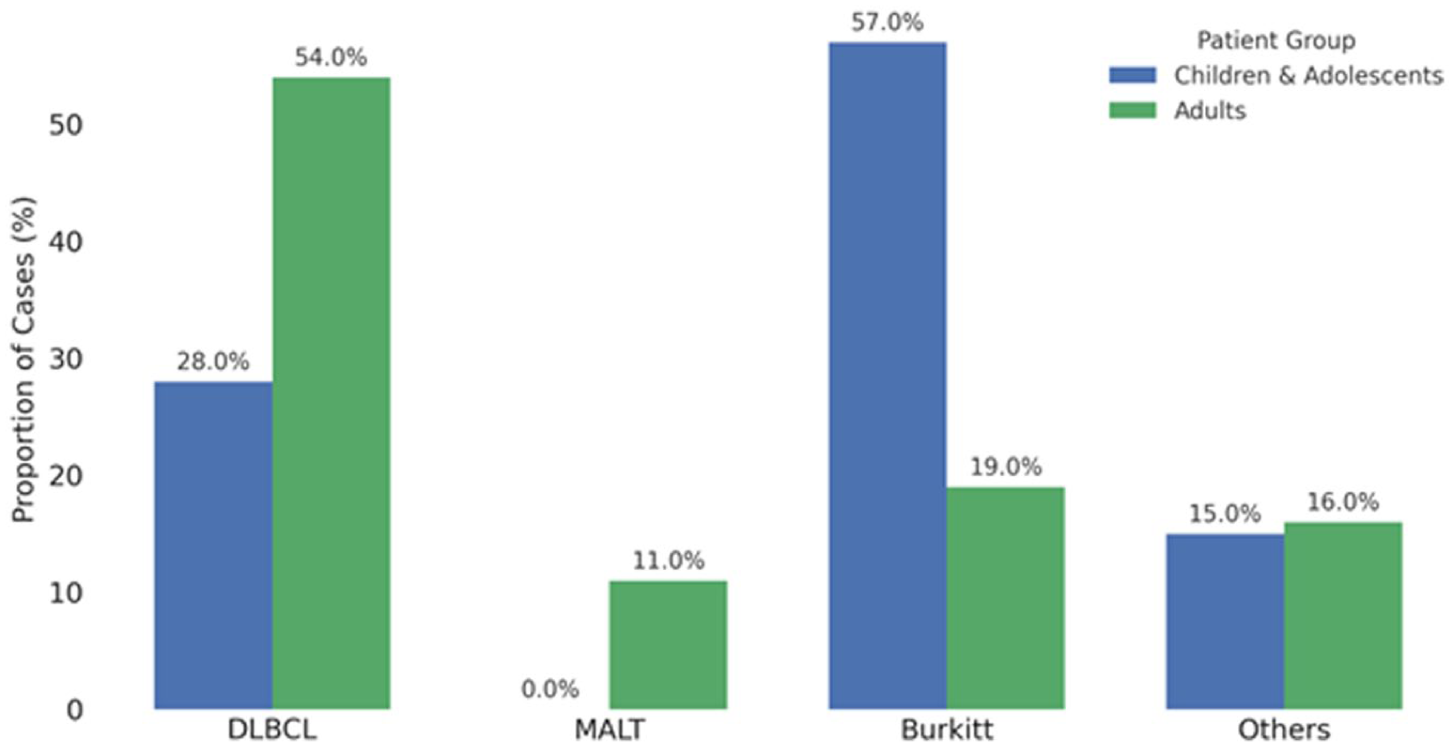
Comparative subtype distribution of gastrointestinal non-Hodgkin lymphomas in adults and children.
Anatomical Distribution of GI Burkitt Lymphoma
The ileocecal region bears an inappropriate burden of the disease. 22 In sporadic gastrointestinal Burkitt lymphoma, ileocecal and distal small bowel (especially ileum and jejunum) are the commonest primary sites- usually with bulky masses, thickened walls, or luminal obstruction on radiography. 25 Another accepted site is the stomach, although far less frequent in classic BL than in other GI lymphomas, but have been reported. 26 BL can also occur in non-typical locations of GI. Pancreatic involvement (primary or secondary) is almost unknown but has been reported as cases resemble pancreatic adenocarcinoma or may even take the form of acute pancreatitis. 6 Primary esophageal BL is nearly unknown, having only a few isolated reports. 27 The findings of the anatomical distribution of GI burkitt lymphomas based on an analysis of the previously mentioned Indian retrospective series of primary 152 GI Lymphomas in all ages is described in Figure 6. In this case report, the anatomy technically does follow the distribution of small intestine as described, however the typical presenting signs of pediatric GI lymphomas of intestinal obstruction and intussusception have not been followed as patterns recognized by a study done solely on primary pediatric GI lymphomas. 28 Although the ileocecal and small bowel segments are the predominant sites of most GI Burkitt lymphomas, the occurrence in the ampullary region with GI bleed in this case is a noncanonical presentation that will demonstrate the spectrum and diagnostic dilemma of BL in the GI tract. Such a localization is strange and emphasizes the reality that one should be clinically suspicious and come up with a conclusive diagnosis of the tissues irrespective of the unexpected locations.

Anatomical distribution of GI Burkitt lymphoma.
Rarity of Pediatric Ampullary Involvement
Primary ampullary involvement by BL in children is exceedingly rare, with only isolated case reports described in the literature.6,16-19 As discussed earlier, pediatric GI Burkitt lymphoma typically arises in the ileocecal region or distal small bowel, making periampullary localization distinctly unusual.22,25,28 In contrast, periampullary masses in adults are relatively common and are predominantly epithelial in origin, most often pancreatic ductal or ampullary adenocarcinoma, which follow a surgically driven management paradigm.29,30 Adult ampullary lymphomas themselves are uncommon and are usually of diffuse large B-cell histology rather than Burkitt subtype.20,21 This case therefore represents an exceptional pediatric presentation, underscoring both the rarity of primary ampullary BL and the diagnostic challenges it poses when compared with more typical adult periampullary tumors.
Treatment, Outcomes, and Prognosis
In pediatric Burkitt’s lymphoma, treatment of the disease is based on short, high-intensity multi-agent chemotherapy schemes such as LMB (Lymphomes Malins B), BFM-style (Berlin-Frankfurt-Münster), R-CODOX-M/R-IVAC, or DA-EPOCH-R (dose-adjusted etoposide, prednisone, vincristine, cyclophosphamide, doxorubicin, rituximab) as adult comparators, combined with systemic and intrathecal CNS prophylaxis to avert leptomeningeal relapse. These regimens achieve 80% cure rates in children and adolescents when delivered promptly with aggressive tumor-lysis prevention. 13
Other landmark trials such as FAB/LMB96—now frequently harmonized with COG ANHL01P1—demonstrated that children with CNS-positive disease and all other risk groups can achieve long-term event-free and overall survival rates of approximately 80% to 85%. 11 Conversely, adenocarcinomas are mainly treated surgically (mostly with curative intent) and show markedly lower prognosis: for example, curative resection of duodenal adenocarcinoma yields a 5-year survival of about 46%, compared with 1% in patients receiving only palliative treatment. 30 The outcome after resection of pancreatic ductal adenocarcinoma remains dismal, with long-term survival of just around 18% in most series. 29
Accordingly, in contrast to pediatric BL which is largely curable when treated expeditiously with intensive chemotherapy, the prognosis of adenocarcinomas is considerably worse even after surgical intervention. Altogether, BL is a chemosensitive and potentially curable malignancy. Early, limited surgical decompression is justified in selected gastrointestinal presentations to prevent cholangitis, reverse hyperbilirubinemia that would delay chemotherapy, and treat bulky disease unlikely to regress fast enough to avoid complications such as perforation, obstruction, or bleeding.12,13
This principle was reflected in this case, where the child underwent a Roux-en-Y choledochojejunostomy to relieve obstructive jaundice (total bilirubin 7.7 mg/dL) caused by an ampullary mass. Surgery in this instance served not as definitive therapy but as supportive, enabling intervention. Although endoscopic retrograde cholangiopancreatography (ERCP) with biliary stenting and percutaneous transhepatic cholangiography (PTC) are generally preferred first-line modalities for malignant biliary obstruction,9,10 several case-specific factors favored surgical Roux-en-Y choledochojejunostomy in this case. Endoscopy demonstrated a large, ulcerated, and friable ampullary mass with active bleeding, rendering ampullary cannulation and sphincterotomy during ERCP particularly hazardous due to the high risk of uncontrollable hemorrhage, a concern also highlighted in reported cases of periampullary BL presenting with bleeding. 6 In addition, pediatric ERCP is technically challenging in the presence of distorted ampullary anatomy and bulky periampullary tumors, with higher failure and complication rates reported in such settings. 16 PTC was likewise considered suboptimal because of the risks of catheter-related infection, bile leakage, and potential tumor seeding, particularly in an immunocompromised child requiring imminent intensive chemotherapy. 12 Under these circumstances, definitive surgical biliary bypass provided reliable decompression, minimized bleeding risk, prevented cholangitis, and enabled timely initiation of curative multi-agent immunochemotherapy, consistent with published pediatric and adult lymphoma experience.7,13
Thus, the overall prognosis in sporadic BL depends primarily on the timely administration of multi-agent chemotherapy, while surgical procedures remain ancillary and directed at overcoming complications that might otherwise delay systemic treatment. Long-term outcomes are excellent in pediatric populations as discussed previously. 13
Conclusion
This report highlights the unusual ampullary presentation of Burkitt lymphoma in a pediatric patient, a site rarely described even within the abdominal predominance of sporadic disease. The case underscores how gastrointestinal BL can masquerade as adenocarcinoma and present with complications such as obstructive jaundice and upper gastrointestinal bleeding, emphasizing the importance of timely biopsy and multidisciplinary management. While intensive rituximab-based chemotherapy achieves cure rates exceeding 80% in well-resourced settings, survival in low- and middle-income countries remains significantly lower. For example, an Indian multicenter cohort of 265 BL patients reported a 3-year event-free survival of 58% and overall survival of 66% due to delays, treatment-related mortality, and limited supportive care. 30 Similarly, a Pakistani pediatric B-cell NHL series documented a 4-year overall survival of 67.1%, reflecting the impact of constrained oncology infrastructure. 3
Future priorities include establishing population-level cancer registries and performing nationwide retrospective analyses to clarify the true burden and outcomes of pediatric BL in South Asia. Investment in early diagnostic pathways, timely referral systems, and capacity-building in oncology and supportive care will be critical to improve survival. Our case contributes to the literature by expanding the recognized anatomical spectrum of BL, while simultaneously highlighting the urgent need for region-specific data and health system strengthening in Pakistan and the wider subcontinent.
Limitations
The primary limitations of this case include the inability to provide histopathologic and immunohistochemical photomicrographs for visual documentation and the absence of preoperative and post operative Positron Emission Tomography-Computed Tomography (PET-CT) imaging. Departmental policies at the originating institution did not allow release of histopathological, ultrasonography or radiological imaging for publication. An exception was made in CT. Although all necessary diagnostic tests were performed and confirmatory evidence for MYC rearrangement was obtained, lack of accompanying images restricted the visual demonstration of histologic features for the readership. Post-operative PET-CT and detailed chemotherapy records following inter-facility transfer were not obtainable, precluding radiographic response evaluation within the early (⩽2-month) post-surgical interval. Nonetheless, the comprehensive immunophenotypic, cytogenetic, and molecular data allowed for a conclusive diagnosis of Burkitt’s lymphoma and reliable exclusion of other high-grade B-cell lymphomas such as DLBCL. We refrain from inferring outcomes beyond the data available.
Supplemental Material
sj-docx-1-icr-10.1177_11795476261429279 – Supplemental material for Unusual Ampullary Presentation of Pediatric Burkitt Lymphoma: Case Report and Literature Review
Supplemental material, sj-docx-1-icr-10.1177_11795476261429279 for Unusual Ampullary Presentation of Pediatric Burkitt Lymphoma: Case Report and Literature Review by Khadija Malik, Muddassir Syed Saleem, Hammad Amjad, Nidal Bin Kamran, Syed Muhammad Faiq Hussain, Umme Roman Akhtar, Abdul Haseeb, Ahmed Asad Raza, Muhammad Areeb Jawed, Abedin Samadi and Adil Ahmed in Clinical Medicine Insights: Case Reports
Footnotes
Acknowledgements
The authors would like to acknowledge the staff and doctors of Civil Hospital Karachi for their support in managing the patient.
ORCID iDs
Ethical Considerations
The study was conducted in accordance with ethical standards and institutional guidelines.
Consent to Participate
Informed written consent was obtained from the patient’s legal guardian in both Urdu (national language) and English. The consent forms have been made available to the editor upon request.
Consent for Publication
Informed written consent was taken from the patient’s legal guardian in the national language Urdu as well as English and the forms have been made available to the editor.
Author Contributions
K.M. and S.M.F.H. contributed to case investigation, methodology, and data resources. K.M. also assisted in conceptualization and validation. M.S.S. and H.A. contributed to conceptualization, methodology, project administration, supervision, visualization, writing of the original draft, revisions and validation. N.B.K. contributed to project administration, drafting, and validation. S.M.F.H. contributed to investigation, methodology, resources, drafting, visualization, and validation. U.R.A. and A.H. contributed to writing the original draft, revisions, and validation. A.J. contributed to data resources and validation. A.A.R, A.A. and A.S. contributed to writing, editing of the final review, and validation. All authors reviewed the manuscript and approved the final version.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data Availability Statement
Not applicable.
Supplemental Material
Supplemental material for this article is available online.