
Abstract
Background:
Inflammatory myofibroblastic tumor (IMT) is a rare mesenchymal neoplasm of intermediate malignant potential that predominantly affects children and young adults. Although pulmonary involvement is most frequently reported, mesenteric IMT is uncommon in pediatric patients. Its diagnosis remains challenging due to nonspecific clinical manifestations and significant radiologic and histopathologic overlap with other mesenchymal tumors.
Case Presentation:
We describe the case of a 5-year-old girl who presented with a three-month history of progressive abdominal distension, anorexia, and weight loss. Radiologic evaluation revealed a calcified mesenteric mass initially suggestive of a calcified fibrous tumor. The patient subsequently underwent exploratory laparotomy with complete surgical excision of the lesion. Histopathologic examination, supported by immunohistochemical analysis, established the diagnosis of inflammatory myofibroblastic tumor. The postoperative course was uneventful, and no evidence of recurrence was observed at the six-month follow-up.
Conclusion:
Although rare, pediatric mesenteric IMT should be considered in the differential diagnosis of abdominal masses in children. The absence of distinctive clinical and radiologic features frequently limits accurate preoperative diagnosis, highlighting the importance of a multidisciplinary approach. Definitive diagnosis relies on histopathologic evaluation and immunohistochemical studies, including anaplastic lymphoma kinase (ALK) assessment. Complete surgical excision remains the cornerstone of management and is generally associated with favorable outcomes. This case emphasizes the need for heightened clinical suspicion and underscores the critical role of immunohistochemistry in establishing an accurate diagnosis and guiding appropriate management.
Keywords
Introduction
Inflammatory myofibroblastic tumor (IMT) is a rare mesenchymal neoplasm of intermediate malignant potential that predominantly affects children and adolescents. 1 Historically described under various terms, including inflammatory pseudotumor, advances in molecular pathology have established IMT as a distinct neoplastic entity characterized by myofibroblastic spindle cell proliferation accompanied by a variable inflammatory infiltrate of lymphocytes, plasma cells, and eosinophils. Reflecting its capacity for local recurrence and rare metastasis, the 2020 World Health Organization (WHO) classification designates IMT as an intermediate-grade soft-tissue tumor and discourages the use of outdated terminology.2,3
IMT may arise in virtually any anatomic location. Pulmonary involvement is more common in adults, whereas extrapulmonary IMT is more frequently observed in younger patients.4,5 In children, the abdomen and pelvis are the most commonly affected sites, followed by the head and neck and thoracic cavities, with additional reports describing involvement of the genitourinary tract, peripheral soft tissues, bones, and the central nervous system.6-8 Intra-abdominal IMT is relatively uncommon, with mesenteric IMT being particularly rare in children. These tumors are difficult to diagnose due to their non-specific clinical symptoms and imaging resemblance to more aggressive neoplasms, such as gastrointestinal stromal tumors (GIST), sarcoma, lymphoma, and other pediatric abdominal malignancies. 9 Imaging findings such as heterogeneous enhancement, internal calcification, myxoid components, and fibrous stroma lack specificity, frequently leading to misclassification prior to surgical intervention.9-11
Given these diagnostic limitations, histopathologic examination with immunohistochemical correlation remains essential for definitive diagnosis. Complete surgical excision is the treatment of choice for localized disease and is generally associated with excellent long-term survival; however, mesenteric involvement carries a higher risk of local recurrence, particularly following incomplete resection.12-14 Owing to the rarity of pediatric mesenteric IMT and the diagnostic uncertainty associated with its presentation, reporting well-documented cases is crucial to improving recognition and management. We herein present a case of mesenteric IMT in a 5-year-old girl, initially misinterpreted radiologically as a calcified fibrous tumor, highlighting the diagnostic challenges and the pivotal role of histopathology and immunohistochemistry.
Case Presentation
A 5-year-old girl presented with a 3-month history of progressively increasing abdominal distension. The swelling initially appeared as a small mass in the upper abdomen and gradually increased in size over time. She also experienced intermittent abdominal pain, reduced appetite, intermittent fever, and significant unintentional weight loss during the same period. There was no history of trauma, vomiting, jaundice, altered bowel habits, or previous abdominal surgery. On admission, the patient appeared well and was hemodynamically stable. Vital signs and anthropometric measurements were within normal limits. There was no pallor, cyanosis, or peripheral lymphadenopathy. Cardiovascular and respiratory examinations were unremarkable, with normal heart sounds and clear bilateral air entry. Abdominal examination revealed a firm, irregular, mobile mass measuring approximately 10 cm × 8 cm in the mid-abdomen, located superior to the umbilicus. The mass was non-tender and not adherent to the overlying skin. No hepatosplenomegaly or ascites was noted. Neurological and musculoskeletal examinations were normal.
Baseline laboratory tests revealed a white blood cell count was within normal range. Renal function tests, liver function tests, and serum electrolytes were within normal limits. Serum LDH was elevated at 908 U/L. Abdominal ultrasonography revealed a large heterogeneously hypoechoic intra-abdominal mass measuring 8.5 cm × 6.9 cm, attached to the peritoneum with absent internal color doppler flow and internal echogenic focus with shadowing (calcification). No hepatobiliary abnormalities, lymphadenopathy, or free fluid were detected. The initial differential diagnosis included a calcified hematoma and, less likely, a desmoid tumor. Subsequent contrast-enhanced computed tomography revealed a well-defined hypodense mass measuring 9 cm × 8.5 cm × 6.1 cm in the mid-abdomen, located to the right of the midline. The lesion demonstrated prominent central calcification with a peripheral enhancing mesenteric soft-tissue component and displacement of adjacent small-bowel loops. The radiologic impression favored a right-sided mesenteric soft-tissue mass with internal calcification, most consistent with a calcified fibrous tumor of the mesentery (Figure 1).
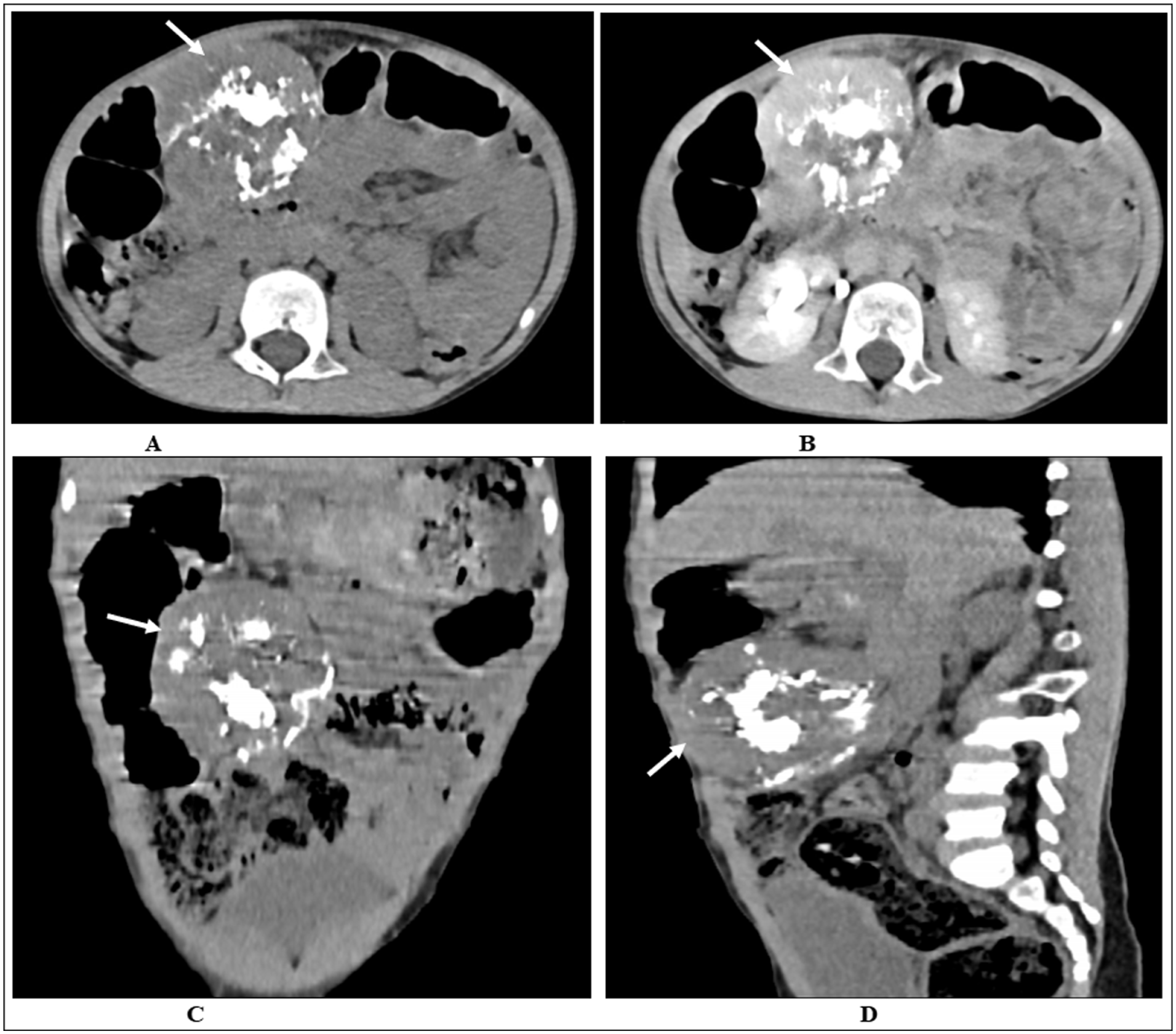
Contrast-enhanced CT scan of the abdomen in axial (A & B), coronal (C), and sagittal (D) planes showing a well-defined hypodense mid-abdominal mass (white arrows; 9 cm × 8.5 cm × 6.1 cm) located to the right of the midline, with central calcification, peripheral enhancing mesenteric soft tissue, and displacement of adjacent small-bowel loops.
Based on the preoperative radiologic diagnosis, the patient underwent surgical excision of the mass. Exploratory laparotomy revealed an encapsulated tumor measuring approximately 9.5 cm × 8 cm, arising from the mesentery approximately 12 cm proximal to the terminal ileum (Figure 2). The mass extended along the mesentery but remained clearly separate from the bowel lumen and exhibited a firm to hard consistency. Complete excision was achieved while preserving the adjacent bowel and surrounding structures. The small bowel appeared normal, and no ascites was present. Several enlarged adjacent mesenteric lymph nodes were identified and biopsied.
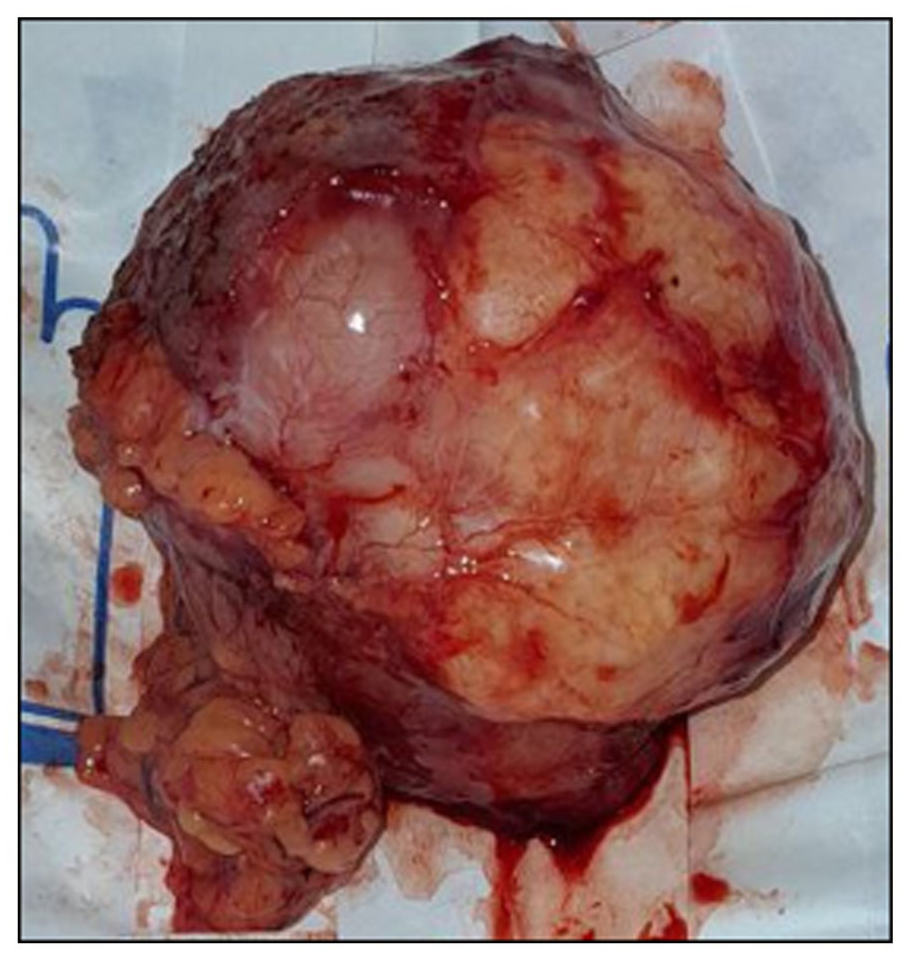
Encapsulated, lobulated, grayish-brown multinodular tumor mass measuring 9.5 cm × 8 cm. The cut surface appears myxoid with focal areas of hemorrhage.
Histopathologic examination demonstrated a relatively bland spindle cell proliferation admixed with inflammatory cells, along with extensive areas of hypocellular hyalinized stroma, calcification, and osseous metaplasia (Figure 3A and B). The sampled mesenteric lymph nodes showed reactive hyperplasia with preserved architecture. The initial histologic impression favored a spindle cell neoplasm consistent with inflammatory myofibroblastic tumor, with immunohistochemical correlation recommended for definitive diagnosis. Immunohistochemistry revealed diffuse cytoplasmic positivity for ALK (Anaplastic lymphoma kinase) and strong diffuse positivity for vimentin (Figure 4A and B). Tumor cells were negative for desmin and CD117. These findings confirmed the diagnosis of mesenteric inflammatory myofibroblastic tumor and excluded other spindle cell neoplasms, including gastrointestinal stromal tumor. Surgical margins were free of tumor, indicating complete excision. The patient did not receive adjuvant therapy. Her postoperative recovery was uneventful, and she was enrolled in a structured surveillance program that included regular clinical assessments and imaging. Abdominal ultrasound was performed monthly, while contrast-enhanced CT of the abdomen was obtained every 6 months. At 6 months, CT imaging showed no evidence of residual disease or recurrence, and the patient remained completely asymptomatic (Table 1).

(A) Gross Pathology: Well-circumscribed, firm, nodular mass (9.5 cm × 8 cm × 8.5 cm) with irregular tan-brown surface and focal yellow areas. Cut surface is tan-white with interspersed yellow regions; solid, without cystic change or hemorrhage and (B) microscopic examination showing spindle-shaped tumor cells within myxoid and collagenous stroma, accompanied by prominent inflammatory infiltrates (hematoxylin and eosin, 100×).

(A) Immunohistochemical (IHC) staining shows diffuse expression of ALK in tumor cells and (B) vimentin is diffusely positive in the tumor cells.
Chronological Timeline of the Case.

Discussion
Inflammatory myofibroblastic tumor (IMT) is an uncommon mesenchymal neoplasm characterized by myofibroblastic spindle cell proliferation accompanied by a prominent inflammatory infiltrate. 8 Although rare, IMT represents an important diagnostic consideration in pediatric patients presenting with abdominal masses, particularly because its nonspecific clinical and radiologic features frequently mimic both benign and malignant intra-abdominal tumors. 9 Mesenteric IMTs are especially uncommon, and their rarity contributes to delayed or incorrect preoperative diagnosis. IMT occurs across a wide age range but most frequently affects children and young adults, with a reported mean age of approximately 9.7 years and a slight female predominance,5,15 consistent with the present case. This case adds to the limited pediatric literature and highlights the diagnostic complexity associated with mesenteric IMT (Table 2).
Comparison of Clinical Features of this Case with General Pediatric Mesenteric IMT.

Clinically, IMT should be included in the differential diagnosis of pediatric abdominal masses, which encompasses spindle cell sarcomas, peripheral nerve sheath tumors, gastrointestinal stromal tumors, lymphomas, inflammatory fibroid polyps, calcified fibrous tumors, and other fibro-inflammatory or pseudo-neoplastic processes. 24 Mesenteric IMTs typically present with nonspecific symptoms such as abdominal distension, pain, anorexia, and weight loss. 20 Constitutional symptoms, including fever, anemia, and weight loss, may occur as a result of cytokine-mediated systemic inflammation.2,17,25,26 These nonspecific manifestations often broaden the differential diagnosis and contribute to delayed recognition (Table 2).
The pathogenesis of IMT remains incompletely understood. Proposed mechanisms include inflammatory or autoimmune triggers, prior trauma, and infectious agents such as Epstein–Barr virus, particularly in selected anatomic sites. 27 However, the identification of chromosomal rearrangements involving the ALK gene on chromosome 2p23 in approximately 50% to 60% of cases supports the neoplastic nature of IMT.28,29 ALK expression is more common in pediatric tumors and has important diagnostic and therapeutic implications. The WHO classification accordingly categorizes IMT as a neoplasm of intermediate biologic potential.2,3
Radiologic evaluation of mesenteric IMTs is often challenging due to the lack of pathognomonic features. On ultrasonography and CT, IMTs may appear as well-defined or infiltrative masses with variable enhancement patterns, fibrous or myxoid components, and occasional calcification.9,20,21 In the present case, ultrasonography and computed tomography revealed a well-defined mesenteric mass with dense central calcification and peripheral soft-tissue enhancement, findings that overlap with those of GISTs, sarcomas, fibromatosis, and calcified fibrous tumors. This overlap underscores that imaging findings alone are insufficient to reliably distinguish IMT from other mesenteric tumors. Ultrasound, as a non-invasive, radiation-free first-line modality, allows real-time assessment of mass morphology and vascularity, while Contrast-enhanced ultrasound (CEUS) can further enhance lesion characterization by delineating microvascular perfusion patterns, although data specific to pediatric IMTs are limited; CEUS has shown utility in improving preoperative diagnostic confidence in abdominal soft-tissue tumors.13,30 Computed tomography (CT) remains essential for detailed assessment of lesion density, enhancement, internal calcifications, and relationships to adjacent structures, which is particularly useful for surgical planning. Magnetic resonance imaging (MRI) offers superior soft-tissue contrast without ionizing radiation and can provide detailed evaluation of tumor extent, margins, and internal composition. While neither CT nor MRI can definitively distinguish IMTs from malignant neoplasms, a multimodal imaging approach beginning with US and incorporating CEUS, CT, or MRI as indicated ensures comprehensive preoperative evaluation.12,13
Histopathologic examination remains the diagnostic gold standard for IMT. Microscopically, IMT is characterized by spindle-shaped myofibroblasts arranged in fascicular or storiform patterns, accompanied by a dense inflammatory infiltrate. 29 Given the substantial morphologic overlap with other spindle cell neoplasms, immunohistochemistry is essential for accurate classification. Tumor cells commonly express ALK, vimentin, smooth muscle actin, and desmin, with variable cytokeratin positivity, while CD117 and CD34 are typically negative, aiding distinction from GIST. 29 Immunohistochemical staining was performed using antibodies against ALK (clone D5F3, Ventana), vimentin (clone V9, Dako), desmin (clone D33, Dako), and CD117 (polyclonal, Dako), according to the manufacturers’ protocols. In the present case, diffuse ALK positivity and vimentin expression, together with CD117 negativity, confirmed the diagnosis of IMT and excluded other mesenchymal tumors.
Complete surgical excision with negative margins remains the standard treatment for localized IMT and is associated with favorable outcomes. 22 Recurrence rates range from 15% to 37% and are higher in extrapulmonary and mesenteric tumors, especially following incomplete resection. 31 In this patient, complete excision was achieved, and no recurrence was observed during follow-up. Although adjuvant therapy was not required, targeted ALK inhibitors such as crizotinib have shown efficacy in unresectable, recurrent, or metastatic ALK-positive IMT and represent a major therapeutic advance.31,32 However, their high cost and limited availability restrict use in resource-constrained settings. In pediatric patients with positive surgical margins, re-excision is preferred when feasible. For cases in which further surgery is not possible, close observation may be appropriate in selected low-risk patients.33,34 Systemic corticosteroids and ALK-targeted therapy may be considered for residual or unresectable disease, while chemotherapy and radiotherapy are reserved for refractory or aggressive cases.33-35 Long-term surveillance is essential due to the risk of recurrence, even after complete resection.
Postoperative follow-up in pediatric mesenteric inflammatory myofibroblastic tumor (IMT) requires balancing the early detection of recurrence with minimizing radiation exposure. Ultrasound (US) is the preferred first-line modality due to its non-invasive nature and ability to provide real-time assessment of lesion size and vascularity. When available, contrast-enhanced ultrasound (CEUS) can further improve lesion characterization by visualizing microvascular perfusion without exposing the patient to ionizing radiation. Magnetic resonance imaging (MRI), offering excellent soft-tissue contrast and no ionizing radiation, is ideal for periodic surveillance, particularly for children requiring long-term monitoring. Computed tomography (CT) should be reserved for situations in which US or MRI findings are inconclusive, urgent cross-sectional detail is required, or complex anatomical assessment is necessary. Prioritizing US and MRI over CT aligns with pediatric radiation safety principles while enabling effective long-term monitoring and early identification of recurrence. A practical, radiation-conscious surveillance strategy involves frequent US during the early postoperative period, periodic MRI for detailed follow-up, and selective CT only when clinically indicated. Suggested intervals based on current literature include monthly US for the first 6 months, followed by MRI every 3 to 6 months over the subsequent 18 months, with continued imaging tailored to the individual patient’s risk profile and clinical course. This approach ensures timely detection of recurrence while minimizing cumulative radiation exposure. 30 In our case, the patient was enrolled in a structured follow-up program comprising regular clinical assessments and imaging. Abdominal ultrasonography was performed monthly, and contrast-enhanced CT was obtained every 6 months. To date, imaging has shown no evidence of residual disease or recurrence, and the patient has remained clinically well. Long-term surveillance remains essential due to the potential risk of recurrence and other complications associated with visceral IMT.
From the caregiver’s perspective, the gradual progression of abdominal symptoms had caused considerable anxiety during the diagnostic period. Following successful surgical treatment, the caregiver reported reassurance and satisfaction with the child’s smooth postoperative recovery.
Conclusion
Pediatric mesenteric inflammatory myofibroblastic tumor (IMT) is an uncommon yet clinically significant entity that often mimics other mesenchymal neoplasms. Its nonspecific clinical presentation and radiologic findings, coupled with histologic heterogeneity, contribute to considerable preoperative diagnostic uncertainty. As highlighted in this case, accurate diagnosis requires a multidisciplinary approach that integrates careful clinical evaluation, detailed imaging studies, histopathologic examination, and immunohistochemical analysis, particularly assessment of anaplastic lymphoma kinase (ALK) expression, to reliably distinguish IMT from other spindle cell tumors, including gastrointestinal stromal tumors (GIST). Complete surgical excision remains the cornerstone of management and is generally associated with favorable short-term outcomes. However, given the recognized potential for local recurrence, sustained long-term follow-up is essential to ensure optimal patient outcomes. This case underscores the importance of maintaining a high index of suspicion for IMT in children presenting with abdominal masses, as timely recognition and appropriate multidisciplinary management are critical to improving prognosis.
Footnotes
Acknowledgements
We would like to thank the family for their consent and cooperation. We would also like to thank all clinicians involved in the child’s clinical care.
Abbreviations
Ethical Considerations
The institution does not require ethical approval for the publication of a single case report.
Consent for Publication
Written informed consent was obtained from the patient’s legal guardian for publication of this case report and any accompanying images. A copy of the written consent is available for review by the Editor-in-Chief of this journal.
Author Contributions
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data Availability Statement
All data sets supporting the conclusions of this case report are available from the corresponding author upon reasonable request.*